 Figure 1 The Probability Cross-sections of the
ns, np and nd Orbitals of Hydrogen
Figure 1 The Probability Cross-sections of the
ns, np and nd Orbitals of HydrogenMany Electron Theory of the Periodic Table
The Azimuthal Quantum Number
In the spectra of all other Elements, the sequence of bands corresponding to the principal quantum number, n was again detectable but the nested bands described by the azimuthal quantum numbers were now split into their separate patterns. The Schrödinger model identifies this angle as the “declination” of the orbital away from the vertical direction. Since this declination cannot matter in a spherical orbital, any orbital labelled with ℓ > 0 must be non-spherical.
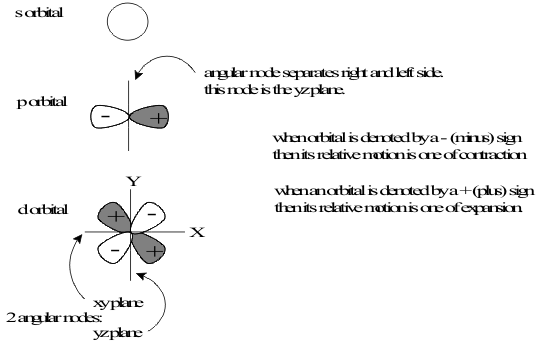
In the spherical harmonic wave functions which solve the Schrödinger equation, (2.3), the spherical shape of the “fundamental harmonic” is converted into successively less spherical shapes, represented by increasing values of ℓ, by splitting the sphere into subspaces with flat planes called “Angular Nodes”.The empirical azimuthal identification labels from atomic spectra coincided with these theoretical values of ℓ as calculated in Table 1.
Table 1 Azimuthal Quantum Labels
|
Empirical Label |
Theoretical Label ℓ = ? |
Number of Angular Nodes |
Number of Lobes ( 2ℓ + 1 ) |
|
s |
0 |
0 |
1 |
|
p |
1 |
1 |
3 |
|
d |
2 |
2 |
5 |
|
f |
3 |
3 |
7 |
|
g |
4 |
4 |
9 |
The Magnetic Quantum Number
In the spectra of all Elements, the transitions identified by the principal quantum number all separated into nested sets of transitions at different energies, depending on the strength of the magnetic field. Since the magnetic field points in one direction, this meant that all the orbitals which were not spherical had been energetically sorted according to their shapes. For example, in the orbital with ℓ = 1, the single flat plane which splits the sphere in half, called the angular node, can be set up in three “orthogonal” directions at right angles to each other, xz, yz and xy. Since none of these is special, the Principle of Uncertainty dictates that all three of these “lobes” must exist together. Geometrically, the sphere can be split in half by the nodal plane to give respectively, front and back, left and right or up and down to give the three independent lobes. The total number of lobes allowed in an orbital is its total number of magnetic quantum number, mℓ values, defining its “degeneracy” as ( 2ℓ + 1 ). The degeneracies for the first five values of ℓ are listed in Table (2.5) and the shapes of the corresponding lobes are shown in Figure 1.
Figure 1 The Probability Cross-sections of the
ns, np and nd Orbitals of HydrogenQuantum Structural Interpretation of Periodic Trends
With the advent of this general quantum description, the opportunity arose of coordinating its quantum number Behaviour labels with the empirical spectroscopy labels. This process is called an “assignment” of the empirical data to the theoretical model.
The most important of these quantum QSARs are those used to Structurally interpret the horizontal, vertical and diagonal Behaviour trends in the Periodic Table. From the 3D spherical polar coordinates in (2.3), this interpretation is split into three parts, the radial part, concerned only with the average radius of the stable electron in any state, the azimuthal angular part concerned with the variations in stability with increasing “declination” of the orbital away from the vertical direction and the magnetic part, concerned with variations in stability with horizontal angle.
Configurations
This one-electron quantum model of atomic orbital structure can be used to understand the Periodic trends if it is used to set up many-electron “configurations” of all the atoms heavier than Hydrogen. To do this, a final electron spin magnetic quantum number, ms must be recognized. This has a value of ½ for each electron. When these electrons are confined to one orbital lobe, the Pauli Principle defines the only allowed interaction as the counteractive pairing of their m s +½ values. In this one way they cancel the magnetic moment of the “filled” lobe. When this pairing Principle is used to fill all of the one-electron orbital lobes needed to hold the number of electrons required by the Atomic Number, Z, of an Element, the order of orbital filling follows the stabilities of the successive orbitals. This process is called the “Aufbau (from the German to “build up”) Principle” and the final set of filled orbital lobes in the neutral atom is called the “configuration” of the atom.
Stabilities of Many-Electron Orbitals
While this Aufbau Principle defines the order of orbital filling as the sequence of orbital stabilities, it does not explain why the orbitals have their particular stabilities. In order to apply the Principal, the reasons for the stabilities of each type of orbital need to be defined. As it turned out, this was a “Nobel Prize” task which, before the advent of computers, required the entire decade of the 1940's to complete the necessary calculations by hand. When they were finished, John Slater was able to extract a very simple set of rules for describing the stability occupied orbitals, which are based directly on the geometries of their spherical harmonic wave functions.
“Slater’s Rules” start with the Structures of the one-electron orbitals described by the Schrödinger wave functions, then take account of the two-electron energies which arise when electrons fill the available lobes in their Pauli pairs. As was seen in the Hydrogen atom, the one-electron orbital energies depend only on the principal quantum number, n. Thus, the most stable configuration of the Hydrogen atom is (1s)1, in which the electron has full access to the nucleus from all directions. In the Helium atom, the Pauli Principle allows the second electron, required in
the neutral atom, to enter the same orbital if it is magnetically “paired”, giving the configuration (1s)2.
The Helium nucleus has two protons but the Helium atom is not twice as stable as the Hydrogen atom because on average in the filled 1s orbital, one of the electrons is always at a smaller radius than the other. Since like charges repel, this closer electron pushes the further electron to a larger average radius, “destabilizing” it within the configuration. Slater called this destabilisation the “shielding” of the nucleus by the closer electron. In effect, the other electron does not have access
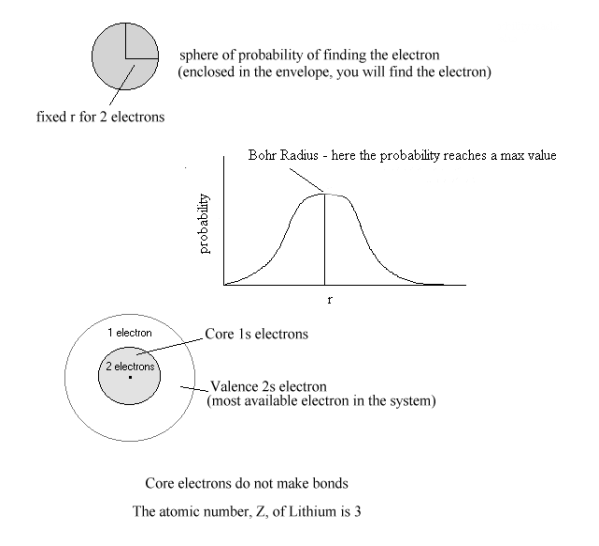 Figure 2 Shielding of Valence Electron of Li
Atom by filled [He] Core
Figure 2 Shielding of Valence Electron of Li
Atom by filled [He] Coreto the full attraction of the two nuclear protons, as shown in Figure (3.1) but is retained in the orbital by an “effective nuclear charge”, Zeff defined as the nominal nuclear charge, reduced by the value of the “shielding constant”, σ, which is derived from Slater’s orbital stability calculations ;
Zeff = ( Z - σ ) (1a)
For the Helium atom, this calculation gives an effective nuclear charge of ;
Zeff = ( 2.0 - 0.3 ) = 1.7 (1b).
which means that each of the two electrons in the ( 1s )2 configuration is attracted by an effective nuclear charge equivalent to 1.7 protons, instead of the full, nominal nuclear charge of 2.0 protons.
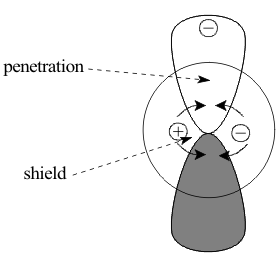 Figure
3 Shielding
of 2p by 2s
Figure
3 Shielding
of 2p by 2sFor simplicity, Slater attempted to extend these radial shielding rules to heavier atoms by assuming that angular nodes do not matter. However, this did not succeed because the angular nodes in orbitals with (ℓ>0) destabilize their electrons by restricting them to angular subspace. These electrons no longer have the same unlimited access to the nucleus from all directions, as they do in s orbitals. Their reduced access increases their shielding constants and the effect can be seen in the decreased effective charge of a 2p orbital of carbon compared to that for a 2s orbital
Zeff ( 2s ) = 3.22 > Zeff ( 2p ) = 3.14 (2)
shown in Figure 3. This increase in shielding gets stronger as the number of angular nodes increases. Calculations in the 1960's by Clementi and Raimondi included the effects of all angular nodes and provided much more accurate values of these shielding constants. Indeed, their work showed that the effects of both the radial and angular contributions to shielding must be used before it is possible to correlate the Behaviour patterns of the Periodic Table to atomic Structure.
Periods
As Slater found, the biggest change in shielding happens when the next electrons added to an atom are forced to occupy an orbital with an increased radius. Therefore, from the Aufbau Principle, the need to start a new Period represents the need to start putting the electron of the next Element into an orbital with the next higher principal quantum number. Then, as Clementi and Raimondi showed, the lowest value of ℓ represents the orbital with the best access to the nucleus and therefore, the Element beginning each new Period always has the configuration ( [n + 1] s )1. Thus, Periods exist because of the radial instabilities described by the principal quantum number, n.
Groups
Along each new Period, this ([n + 1] s) orbital becomes more stable, because each new Element adds a new proton to the nuclear charge. After the second Element in each Period however, this orbital is filled, in the configuration ( [n + 1] s )2 . Because this configuration is the same at the beginning of each Period, it defines the electronic Structure of the Elements in the Alkali Metal or “s” Groups of the Table. The Period can only continue by filling orbitals with higher ℓ values if they are available. This is impossible in the first Period, which can therefore only contain the two first Group Elements.
In all later Periods, the ([n + 1] p) orbitals are available. By the Pauli Principle they represent six additional Elements in the Period. Because the filled configuration ([n + 1] p)6 is the same at the end of each Period, it defines the electronic Structure of the Elements in the Main or “p” Groups of the Table.
However, this p-Group pattern is observed in the next two Periods, in spite of the formal availability of 3d orbitals in the ( n = 3 ) Period. The destabilisation due to the presence of two angular nodes in these 3d orbitals is so great that the ( n = 3 ) Period does not continue past the filling of 3p orbitals by starting to fill the 3d. Instead, this Period ends after filling the (3p)6 configuration and the Period corresponding to filling ( 4s ) begins. This shows that the access to nuclear charge allowed from all directions in the 4s orbital is better than the restricted access available to the nucleus in the doubly divided space of a 3d orbital. In the terms normally used to describe this situation, an electron in the 4s orbital “penetrates” the “shield” formed by the filled (3p)6 configuration better than one the 3d orbital can. Again, by the Pauli Principle, the ten Elements represented by the filling of these ([n - 1] d)10 configurations in each Period defines the electronic Structure of the Elements in the Transition Metal or “d” Groups of the Table.
However, in each successive Transition Metal Element, the increasing number of protons makes the ([n - 1] d) orbitals become more stable, until at the end of the ten Elements, this filled orbital is more stable than the (ns) orbital which defined the start of the Period. Thus, the last two Elements engage in chemical reactions by using the (ns) electrons instead of the ([n - 1] d) electrons. Thus, they form the separate “Noble Metal” Group between the Transition Metals and the Main elements.
In the Period begun with the filling of the (4s) orbital, the (4f) orbital becomes formally available. However, the nuclear access allowed to electrons in this orbital by its three angular nodes is so limited that they cannot penetrate the shield of the filled (4p)6 and even the (5p)6 configurations, until the Elements corresponding to filling the (6s) orbital are reached. Thus, the Rare Earth of f Groups do not appear in the Periodic Table until n = 6. Thus, while the relationships are much more complicated than those involved with Periods, Groups exist because of the angular instabilities described by the azimuthal quantum number ℓ.
Shell Effects
Along each new Period described by [n + 1], within a Group described by an appropriate value of ℓ, the orbital fills with increasing stability until the configuration reaches ( [n + 1] ℓ ) ℓ , called the “half-filled shell”. At the next Element, the configuration stability suddenly decreases. This “glitch” correspond to an increase in two-electron repulsion because the additional electron for the first Element after the half-filled shell must obey the Pauli Principle and form a diamagnetic pair with one of the electrons already present in one of the half-occupied lobes. Thus, Half-filled Shell Effects exist because of the Pauli Pairing instabilities described by the magnetic quantum number mℓ .
Sequences
Empirically, the Diagonal Sequences of Elements, identified by simultaneous increases along Periods and down Groups, have remarkably constant Behaviour, as long as the Sequence stays inside a Block. In terms of Effective Nuclear Charge, the increasing stability achieved by adding an extra proton along a Period is essentially exactly cancelled by the decreasing stability from adding an extra filled set of shielding orbitals down the Group. The most prominent example of this Sequencing Effect is the consistency of Behaviour in the “Semi-metals” from Boron to Astatine but the same Effect can be perceived throughout the Periodic Table. Thus the Diagonal Sequence Effect exists because of the counter-balancing stabilization due to the increasing value of the azimuthal quantum number, ℓ and the destabilisation due to the increasing value of the principal quantum number n.
Chemically Relevant Energy Parameters
Chemical Behaviour and these simplest supporting Structures only exist in a limited range of “chemical temperatures”. The lowest temperature is defined by the absence of any reaction Behaviour and the highest, by the absence of bonding Structures. Since the temperature of a system depends directly on its internal energy, this chemical temperature range can be translated directly into a chemical energy range. Then, since Chemistry concerns reaction Behaviour and the bonding Structures of atoms, it is most convenient to define this chemical energy range in terms of the observable energies of atoms.
In quantitative terms, the low temperature end of this energy range can be defined by the “exoergonic” (energy releasing) atomic Actitivity of capturing an electron. This energy expelled during this “cooling” Behaviour is called the Electron Affinity of the atom. Similarly, the high temperature end can be defined by the “endoergonic” (energy absorbing) Behaviour of having an electron stripped away. The energy absorbed during this “heating” Behaviour is called the Ionization Potential of the atom.
After either of these Behaviour is complete, the amount of energy moved out of or into the atom can be interpreted Structurally. If an atom acquires an electron, the new electron can only be bound into the most stable condition available in the atomic Structure. At the other end of the energy scale, if the atom is forced to give up an electron, it can only come from the least stable condition provided by the atomic Structure.
However, when these atoms are placed into a “condensed phase”, their Behaviour and supporting atomic Structures are affected by the Behaviour and Structures of the neighbouring atoms. This interatomic interaction makes it necessary to redefine both the low and high energy limits of their chemical energy ranges. In these media, the atoms not only attract and attempt to retain their own least stable electrons but may also attract and possibly even capture the least stable electrons from any of its neighbouring atoms. Thus, the forces exerted by the neighbouring atoms of a condensed phase effectively change the low and high temperature stabilities of the atom.
The need for this concept of the Chemical energies of associated atoms was first recognized by Linus Pauling in 1935. By comparing the stabilities of simple salts, he showed that their differences in bond energies could be factored into an empirical atomic property that he called the “Electronegativity”, χ, of the Element. Later, Robert Mulliken defined it theoretically as the net energy of electron retention by associated atoms. This was represented as the averaged sum of their two separate abilities of retaining their own electrons, described by the Ionization Potential and of stealing electrons from surrounding atoms, described by the Electron Affinity of the isolated atom;
χ = ( I.P. + E.A. ) / 2 (3a)
In this sense, the energy defined by Electronegativity corresponds to the force called the “polarizing power” Π, of an associated atom. This Coulombic force can distort or “polarize” the electrical charge on any neighbouring atoms. This polarizing force is derived by dividing the Electronegativity energy by the radius of the atom ;
Π = χ / r (3b)
While this discovery of Electronegatrivity was essential, it was equally obvious that the corresponding tendency for the electrical charge on atoms to distort in the presence of this polarizing force would represent their “polarizability”. However, it required thirty more years of research before it was possible for Ralph Pearson to describe the energy corresponding to this polarizability as the “Hardness”, η, of an atom. Then, in 1990, parallel to Mulliken’s theoretical definition of Electronegativity, Peter Atkins finally defined Hardness as the net energy of electron loss from associated atoms. This was represented as the averaged difference of their two separate abilities of losing their own electrons, described again by the Ionization Potential or of rearranging them internally, described again by the Electron Affinity;
η = ( I.P. - E.A. ) / 2 (4a)
The corresponding polarizability is defined like all types of susceptibilities by the ratio of the volume of the affected atom to its own stabilizing charge, represented by its Hardness ;
α = r / η (4b)
In some applications, it is more convenient to define the lower limit with the inverse polarizability property of associated atoms called the “Softness” of the Element.
In effect, the Hardness of associated atoms represents the change in their lower limit of chemical energies from the Electron Affinity of isolated atoms. Likewise, their Electronegativity represents the change in their upper limit from the Ionization Potential. These changes stabilize the associated atom compared to the isolated atom at both limits. However, the Ionization Potential is a much larger energy than the Electron Affinity. Thus, Electronegativity of the associated atom gives a small stabilization compared to the Ionization Potential of the isolated atom but its Hardness gives a very large increase in its stability compared to its Electron Affinity. The chemical consequences of this become obvious in a detailed look at the Chemistry of the Periodic Table.
Empirical Trends of Chemical Behaviour
The chemical energies of isolated atoms are known from bulk electrical measurements on gases. The chemical energies of associated atoms in isolated molecules are assessed by the theoretical definitions of Electronegativity and Hardness, in equations (3.3b and 3.4b). The energies of atoms in condensed phase environments can be measured electrochemically as Reduction Potentials for atoms accepting electron and as Oxidation Potentials for atoms losing electrons. Since they all represent the fundamental Behaviour of electron loss or gain, any of these definitions of chemical energy, shown in Table 2, can be chosen to act as a measurement of atomic Behaviour and plotted against the Structural Atomic Number, Z, representing the number of protons in the nucleus, to reveal the empirical patterns of Elemental Behaviour in the Periodic Table.
Table 2 Forms of Chemical Energy Behaviour
|
Chemical Energies | |||
|
Behaviour |
Isolated Atom |
Associated Atom |
Condensed Atom |
|
Electron Loss |
Ionization Potential |
Electronegativity |
Oxidation Potential |
|
Electron Gain |
Electron Affinity |
Hardness |
Reduction Potential |
These plots show that the trends in the Chemical energies involved with both the electron
loss and gain Behaviour increase along the horizontal Periods, decrease down the vertical Groups and stay essentially constant in the diagonal Sequences of the Elements. However, there are discontinuous decreases along each Period at the beginning, middle and end of each major Block of Elements, including the Alkali Metals, the Transition Metals, the Lanthanide Metals and the Main or Non-metal Elements. Likewise, there are discontinuous increases in these energies down each Group at every second Period, coinciding with the introduction of each new Block of Elements. Finally, the diagonal Sequences are repeated within each major Block of Elements as shown previously in Figure (1.1). These repetitive Behaviour patterns lead to a naming system for the Periods and Groups of the whole Periodic Table. The Periods were numbered in order, starting with each new Alkali Metal, giving n a range from 1 to ∝. The Groups of the Alkali Metals and the Non-metals in the Short Form of the Table were likewise numbered in order from 1A to 8A. However, the Behaviour of the Elements in the Transition Metal Block so closely resembled those of the Main Block that they were numbered in the same order, from 3B to 8B. Unfortunately, since there are ten Elements in each Period of the Transition Element Block, there were not enough labels. To solve this, the Elements in the Groups headed by iron, Fe, cobalt, Co and nickel, Ni were all put into one Group, labelled 8B. Then, since the remaining two Groups, headed by copper, Cu and zinc, Zn resembled the Alkali Metals, they were labelled 1B and 2B. Recently this descriptive but awkward system was replaced by the simpler one of labelling all long Periods in order, 1 to 18.