Group 6A Elements ; The Chalcogens (“Copper Producers”)
Spatial and Electronic Structures
As in Groups 2A to 5A, the (ns)2 electrons of Group 6A Elements can penetrate the core of filled p orbitals from all directions but as before, this core provides a shield with no defects which makes this valence pair of electrons unstable. However, this shielding is even more effective against the (np)2 electrons which can only penetrate to the nucleus from the two ends of its p lobe. Thus, in general, these (np)2 electrons are more easily removed than the (ns)2 electrons.
However, in Oxygen, the [(1s)2] core provides such especially poor shielding that both types of valence electrons can easily penetrate through to the nucleus and there .is no significant difference in their penetrating power. However the core becomes very effective down the Group in the Elements Sulphur to Tellurium, so the differences in penetration by their valence (ns)2and (np)4 electrons becomes much more pronounced. The net effect of these shielding conditions is that the Effective Nuclear Charge ZEff, begins at moderate values in Nitrogen and decreases down the Group. This leads to a progressive increase in the atomic radius, representing the spatial aspect of atomic Structure of and a decrease in Hardness and Electronegativity, representing the electronic aspects of atomic Structure. The correlation of electronic configurations to spatial and electronic Structures is shown in Table 1
Table 1 Group 6A Correlation of Electron Configuration to Electronic Structure
| Element
Name |
Structural
Configuration
[core electrons] (valence electrons) |
r
nm |
η
kJ/M |
χ
kJ/M |
| Oxygen | [(1s)2] (2s)2 ( 2p)4 | 0.15 | 585 | 725 |
| Sulfur | [(1s)2(2s)2(2p)6] (3s)2 (3p)4 | 0.18 | 400 | 600 |
| Selenium | [(1s)2(2s)2(2p)6(3s)2(3p)6] (4s)2 (4p)4 | 0.19 | 365 | 575 |
| Tellurium | [(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)10(4p)6](5s)2(5p)4 | 0.21 | 324 | 545 |
| Polonium | [(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)10(4p)6(5s)2
(4d)10(5p)6] (6s)2 (6p)4 |
0.23 | 300 | 500 |
Molecular Forms of Group 6A Elements
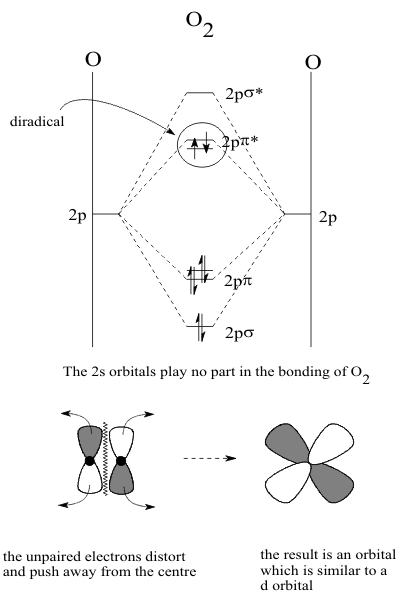
As the Elements increase in Hardness and Electronegativity across the Periodic Table, their bonding progresses from metallic to covalent. While the Rare Gas Rule can be used to define the number of valence pairs available in the atoms of the p-block Elements, it cannot be used to describe why most of the p-block elements in the first Period occur naturally as diatomic gases while their heavier congeners are found as solids with many “allotropic” forms. The LCAO model used to describe the Structures of Group 5A molecules can be applied directly to this question and in particular, it clearly shows the differences between the Structures of Oxygen and Sulfur. Just as in the description of the gaseous diatomic molecular free radical NO, it is only necessary to use the valence (2p)4 orbitals of the O atoms to describe the bonding of the diatomic species O2. The electronic Structure of this compound is described the same way as NO, as shown in Figure 1.
When the Aufbau Principle is applied, the least stable electrons are again placed in the doubly degenerate (2pπ*) orbital. However, since these two orbitals have the same energy, they provide four equivalent positions for these two electrons. This means that the electrons may occupy this orbital as a Pauli pair but can instead each occupy a different lobe as an unpaired electron. Since this option places the electrons farther apart, it is the more stable form of O2 and is thus found in the air as the “diradical”. Since this is an antibonding orbital, it reduces the bond order form 3 to 2. By the Woodward-Hoffman criterion, this places a node between the O atoms so that its Structure resembles that of a (nd) orbital.
This Structure is so stable that this diatomic entity has both Redox and Acid-Base Activities of its own distinct from the Activities of Oxygen atoms combined with other Elements. These specific diatomic Activities can all be described from the electronic Structures defined on the LCAO MO diagram. Using only the (2pπ) and (2pπ*) orbitals of the diagram, the Aufbau Principle is used to define the electronic Structures of the stable reduced and oxidized derivatives of O2 in Figure 2..

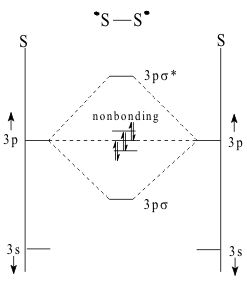
In contrast, the shielding of the (3p) orbitals of sulfur by the filled (2p)6 core, Figure 3, makes the S atom so large that (3pπ) overlap cannot occur.
Thus, in the LCAO MO energy level diagram of S2, shown in Figure 4, the (3pxy) lobes remain non-bonding and the molecule is only stabilized by one (3pzσ)2 bonding pair.
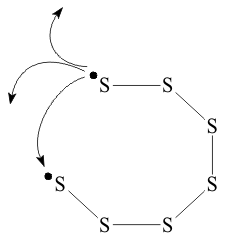
The non-bonding pairs are strongly Lewis basic and stabilized by donationto other species, including by forming σ bonds to other S2 units. When this occurs, the bonds can form in many different ways, producing the many allotropes of elemental S all based on polymers of the (S2) unit ;
X (S2)g ⇒ (S2)x (1)
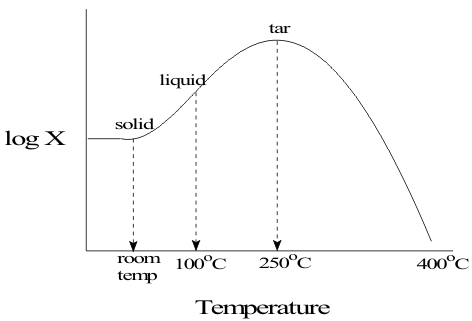
where X can be very large. Cyclic allotropes, such as S6, S8, S10, S12, S16, etc. Shown in Figure .5 persist at low temperatures since by Entropic considerations, it is easier to find the other end of a short polymer than that of another polymer.
However, as the temperature rises to 250°C, these cyclic allotropes convert to linear polymers of increasing length. This increases the viscosity X of the material as shown in Figure 6 until the polymers begin to disintegrate at higher temperatures.
SONs of Group 6A Elements
Since these atoms are usually covalently bound within the Structures of compounds, their Oxidation Numbers again cannot be assigned from experimental measurement of the their charges. Instead, as in earlier Groups, the SONs must again be assigned theoretically on the basis of the relative Electronegativities of the atoms in the bond. Following the Octet Rule, the range of Oxidation Numbers in Group VI is once again limited by the Structures of the previous and the next rare gas respectively. Removing all valence electrons to achieve the configuration of the previous Rare Gas gives an upper SON limit of +VI Adding enough electrons to fill the valence shell and achieve the configuration of the next Rare Gas gives a lower SON limit of -II for the atoms of this Group. Following the Pauli Principle, the intermediate SONs occur when Pauli pairs are preserved. Thus, in Group V these are the one reduced value of -II, and the oxidized values of +II, +IV, +VI.
Electronic Structures of Chalcogen Compounds
When these Rules are applied to Group 6A compounds, apart from Oxygen they again assign SON values over the entire range from the lower to upper Rare Gas limits. Thus, all single bonded Group 6A compounds have the molecular formulas Z2Z’ , Z4Z’ or Z6Z’.
When these Rules are applied to compounds of Oxygen, they identify the value of (SON)Z in compounds with Elements with Electronegativities lower, similar and higher than Oxygen, with an χ value of 1035kJ/M. The formal SON values of Oxygen in compounds across this whole range are given in Table 2..
Table 2 SON Values for Oxygen in X2O Compounds
| Compound | χX | (SON)X | (SON)O |
| Li2O | 290 | +I | -II |
| H2O | 690 | +I | -II |
| OF2 | 1000 | -I | +II |
Reduction to Minimum SONs
In reactions between these Group 6A Elements and less Electronegative metals, Pauling’s Electroneutrality Rule predicts that to minimize the energy of the whole system, the Group 6A atoms would gain electrons into their unfilled (np)4 orbital from the metallic atom. This “reduces” the Group 6A Element to an anion with a minimum SON of -II and “oxidizes” the less Electronegative Element to a cation. For example, in reaction with the Alkali Metals, M, the Group 6A Element, Z would be reduced to form a product ;
2M + Z ⇒ M2Z (2)
Depending on its Polarizing Power, the oxidized species M (+n) may bond to the Group 6A anion, to form either ionic compounds such as lithium oxide, Li2O, or covalent compounds such as water, H2O or hydrogen sulfide, SH2 .
In all of these compounds, the assignment of the minimum SON value of -II to the Group 6A Element again defines this Structure as “electron-rich”. As with Group 5A, in the Structure of X2Z compounds, the Group 6A atom not only brings enough electrons to make the two (X-Y) bond pairs but also brings two additional lone pairs which remain “non-bonding” in the valence shell.
In most cases, χY > χX , so that the Activities of these compounds are dominated by donation of one or both of these extra electron pairs, either as a Lewis base or, in the irreversible reaction limit, as a “reducing agent”. This “Donicity” increases down Group 6A because increasing shielding by the core makes the filled (ns)2(np)6valence shells increasingly unstable. The decrease in χ is now extremely large from O to S but from S to Se, the shielding by the filled (3d)10 shell is very ineffective and the decrease in χ is minimal. Then, from Se to Te, the addition of the second shield of (4d)10 makes a more effective d-orbital shield and the drop in χ is so large that it represents the change from a nonmetal to a metallic Element..
The “Chalconide” Compounds
All of the Group 6A Elements form ZH2 compounds, which, apart from water are identified by the suffix “ide”, connoting ionic bond character. Their Structure and Activity properties are given in Table 3..
Table 3 The Structures and Activities of ZH2 Compounds
| Name | Formula | Bond Stability
SB
(kJ/M) |
Bond Type TB
(Δ kJ/M) |
Bond Angle (degrees) | Acidity
pKA(25C) |
| Water | H2O | 458 | 345 | 104.5 | 15.7 |
| Hydrogen Sulfide | SH2 | 363 | -65 | 92.2 | 6.9 |
| Hydrogen Selenide | SeH2 | 276 | -140 | 91 | 3.8 |
| Hydrogen Telluride | TeH2 | 238 | -240 | 89.5 | 2.6 |
| Hydrogen Polonide | PoH2 | ≈200 | -315 | < 90 | < 2 |
These data show that the difference between the Element and Hydrogen reverses from (H+-Z-) in water to (H--Y+) in SH2 and become increasingly hydridic. Again the (H-Y-H) bond angles decrease and the Bronstead Basicity, measured as the Proton Affinity of these compounds, is weaker in H2 O than in NH3 and becomes insignificant for all of the heavier Element-ides.
Again, because of their importance the Structure and Activity properties have been described by Valence Bond and the Molecular Orbital models. The Valence State Electron Pair Repulsion theory describes water as a molecule with two pair bonds and two lone pairs with hybridization again defined as sp3. If these pairs repel each other equally, this molecule should also be tetrahedral, with a (H-Z-H) bond angle of 109.9o, The difference from 104.5o is again explained by the diffuseness of lone pairs but the weaknesses of VSEPR show up even more clearly with Group VI.
Experimentally, the rapid decrease in the (H-Z-H) bond angle from water to Hydrogen Telluride again shows that the initial sp3 hybridization is inappropriate. The bond angle of less than 90o in Hydrogen Telluride implies that the two bonding pairs only occupy the (6p) orbitals of Te -II. The lone pairs have both apparently disappeared into the electronic core of the Tellurium atom as a filled (6s)2 and a filled (6p)2 orbital which both penetrate into the core. Again, this progressive change in electronic Structure invalidates the assumption of sp3 hybridization and accounts for the weakening of these bonds, the change in bond type and the reduction in Bronstead basicity.
LCAO Description of Bonding
The bonding in H2Z molecules can be described with LCAO MO theory by first recognizing that the H atoms are chemically equivalent in a plane. Depending on their erelative phases the H(1s) AOs can correlate to form either a Totally Symmetric “a1" or an Antisymmetric “b1” Group Orbital. These two GOs can overlap with any of the Z(ns) or Z(np) AOs of appropriate symmetry to form the MOs. In practice, the “a1" GO can overlap with both the Z9ns) and the Z(np)z AOs, while the three “b1” GO can overlap with the Z(np)x AO. The resulting “a1" MOs are like σ MOs, as shown in Figure 7.
The “b1” MO is the Antisymmetric overlap shown in Figure 8 and is like one half of a π set.
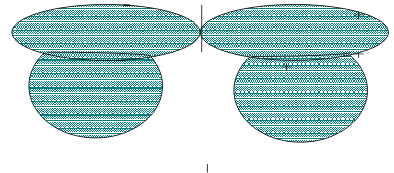
Figure 8 The Antisymmetric b1 Bonding MO of ZH2
According to the Woodward - Hoffman criterion, either phase of the valence (ns) and (np) orbitals can interact simultaneuosly with each of the H(1s) GOs. If the Z(ns) or Z(npz) AOs are in-phase with the H(1s) GOs, the bonding a1 and b1 MOs are formed. If they are out-of -phase, the corresponding a1* and b1* antibonding MOs are formed.
These two types of MO, a1 and b1, achieve different amounts of overlap because of their different geometries. Since the Z(npz)a1 MOs in particular have much greater overlap than either the Z(ns)a1 or the Z(npz)b1 MO, they are more covalently stabilized. The reverse order then occurs for the corresponding antibonding MO energies. As shown in Figure 9, the Energy Level Diagram is constructed the same way as with diatomic MOs. The VSIPs of the Group 6A atom orbitals are plotted on the left vertical axis and the VSIPs of the two degenerate H(1s) orbitals are plotted on the right vertical axis.
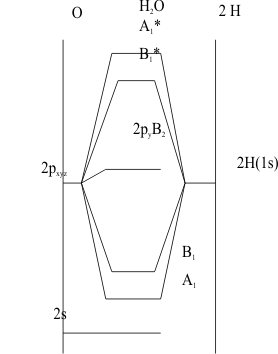
Their two GOs are labelled as a1 and b1. The energies of the polyatomic MOs are then plotted between the vertical axes as stabilized bonding and destabilized antibonding MOs The electronic configuration of the ground state of these molecules is then determined by applying the Aufbau and Pauli Principles. As usual, the available valence electrons from the Group 6A atom and the two H atoms are placed together into the most stable orbitals of the system. Thus, they will first occupy the non-bonding (ns) of the Group 6A atom then, they will fill the two bonding MOs and the final pair must occupy the formally non-bonding (npyb2), which is usually slightly antibonding in real molecules.
The two bonding MOs are made entirely from the two (np) orbitals of the Group 6A atom and are again all that are needed to make a Stable molecule with the equivalent of one bond to each H atom. Again, the fact that the (ns) orbital is unused and that none of the MOs point directly at the H atoms does not matter to the stability of these molecules.
The bond angle is determined by the balance of the overlap in the a1 MO, pulling the angle inwards against the b1 overlap pulling the angle open towards a linear shape. The two lone pairs are very different. One is very stable in the (nsa 1) orbital and is not involved in determining the shape of the molecule at all. The other is isolated in the unused (npyb2) which is very unstable and perpendicular to the molecular plane. This lone pair is donated to electron-deficient molecules, as a polar covalent bond from water or a reducing pair from the heavier congener hydrides..
Oxidation of the Elements
In the reactions of Group 6A Elements with more Electronegative nonmetals, Pauling’s Electroneutrality Rule predicts that, to minimize the energy of the whole system, the Group 6A atoms would lose electrons from either or both of the (ns)2 and (np)4 orbitals to the nonmetallic Element. This “oxidizes” the Group 6A Element to a stable cation of Oxidation Number +II, +IV or +VI, with the Structural parameter values in Table 4, and reduces the nonmetallic Element. In other compounds, the non-metals oxidize these Group 6A Elements to the SON, +IV with the properties given in Table 5 or to +VI with the properties given in Table 6.
Table 4 Electronic Structures of Stable Group 6A Oxidation Number +II
| Stable
ON |
Configuration
[Core](valence) |
r
nm |
η
kJ/M |
(106)α
(nm3)M/kJ |
χ
kJ/M |
Π (10-4)
kJ/Mnm |
| O +II | [ He ](2s)2(2p)2 | 0.02 | 965 | 0.008 | 4350 | 21.8 |
| S +II | [ Ne ](3s)2(3p)2 | 0.05 | 530 | 0.24 | 2800 | 5.6 |
| Se +II | [ Ar ](4s)2(4p)2 | 0.06 | 465 | 0.46 | 2500 | 4.2 |
| Te +II | [ Kr ](5s)2(5p)2 | 0.12 | 500 | 3.45 | 2250 | 1.9 |
| Po +II | [ Xe ](6s)2(6p)2 | 0.12 | - | → | - | - |
Table 5 Electronic Structures of Stable Group 6A Oxidation Number +IV
| Stable
ON |
Configuration
[Core](valence) |
r
nm |
η
kJ/M |
(106)α
(nm3)M/kJ |
χ
kJ/M |
Π (10-4)
kJ/Mnm |
| O +IV | [ He ](2s)2(2p)0 | - | - | - | - | - |
| S +IV | [ Ne ](3s)2(3p)0 | 0.04 | 1250 | 0.05 | 5775 | 14.4 |
| Se +IV | [ Ar ](4s)2(4p)0 | 0.05 | 1225 | 0.1 | 5350 | 10.7 |
| Te +IV | [ Kr ](5s)2(5p)0 | 0.1 | 1025 | 1.00. | 4625 | 4.6 |
| Po +IV | [ Xe ](6s)2(6p)0 | 0.1 | - | - | → | - |
Table 6 Electronic Structures of Stable Group 6A Oxidation Number +VI
| Stable
ON |
Configuration
[Core](valence) |
r
nm |
η
kJ/M |
(106)α
(nm3)M/kJ |
χ
kJ/M |
Π (10-4)
kJ/Mnm |
| O +VI | [ He ](2s)0(2p)0 | - | - | - | - | - |
| S +VI | [ Ne ](3s)0(3p)0 | 0.03 | 9300 | 0.002 | 17800 | 59.3 |
| Se +VI | [ Ar ](4s)0(4p)0 | 0.04 | 3525 | 0.02 | 11400 | 28.5 |
| Te +VI | [ Kr ](5s)0(5p)0 | 0.05 | 3200 | 0.04 | 10000 | 20 |
| Po +VI | [ Xe ](6s)0(6p)0 | - | - | - | - | - |
Bonding Structures of Oxidized Chalcogens
As in the earlier Groups, the type of [ Z-Z’] bond, ionic, donor covalent or covalent, depends on the relative polarizing powers Π and polarizabilities α of the two Elements forming the bond pair. Again the ranges of these parameters make the Group 6A Elements “electronically amphoteric” so that they usually achieve charge satisfaction by covalent sharing rather than by ionic exchange of electron pairs.
In spite of this, it is again very useful to identify the Oxidation Numbers of Group 6A Elements in their compounds in order to describe how electron pairs are rearranged during reactions. The range of Oxidation Numbers is again limited by the Structures of the previous and the next Rare Gas respectively, giving an upper limit of +VI and a lower limit of -II. However, since these atoms are usually covalently bound, their Oxidation Numbers again cannot be assigned from direct experimental measurement of the their charges. Instead, as in earlier Groups, the SONs must be assigned theoretically on the basis of the relative Electronegativities of the atoms in the bond, using the Rules for Stable Oxidation Numbers..
Chalcogen Halides
As with Group 5A Elements, the simplest oxidation reactions occur when the nonmetallic Element acquires the valence electrons of the Group 6A atom, becoming reduced to a stable anion, while the Group 6A atom is oxidized to a cation. In oxidation reactions with the Halogens, Z2 ;
Z + 3Z’2 ⇒ ZZ’6 (3)
and the resulting anion forms single σ bonds to the Group 6A cation. However, the Polarizing Power of Group 6A cations is so high that the Halide ions are forced to provide donor covalent bonds to the oxidized species which results in formation of molecular compounds instead of salts. In general, these compounds have formulas ZZ’2, ZZ’4 or ZZ’6 corresponding to their SONs. However, like the hydrogen compounds, these halide derivatives are electron-rich. For the Z +II cations, in OF2, the poor shielding in O +II cation allows the O (2s) and (2p) valence orbitals to overlap with the a1 GO of the two F - anions to form a bond angle of 101.5o.
However, the much higher shielding in the heavier congeners again stabilizes one of the non-bonding pairs in the (ns) orbital and for Group 6A, it also isolates the other lone pair into the (np)yb2 lobe. At the same time the increase in shielding preferentially stabilizes the (np)z lobe faster that the (np)xb1 orbital. Especially when the Electronegativity of X is high, this energy separation reduces the (X-Z-X) bond angle, down to 98o in SF2, and increases the Lewis basicity of the (np)yb2 lone pair.
In the Z +IV cations, the higher central charge increases this separation in the energies of the (ns) and (np) orbitals even further. At the same time the lone pair in the (np)yb2 MO is replaced by a bonding pair. However, by a Quantum Mechanical Rule called the “Jahn-Teller Theorem”, the (np)xb1 and (np)yb2 bonding orbitals cannot be degenerate meaning that ZZ’4 molecules cannot be flat. Thus in pyramidal SF4, the two pairs of (S-F) bonds have different bond lengths and angles.
For the Z +VI cations, the separation of (ns) and (np) valence orbital energies increases even further, bringing the empty (np) orbitals close to the energy level of the empty (nd) orbitals in the heavy cations. Thus each all of these electron deficient orbitals can accept bonding or non-bonding pairs, expanding the valence beyond the number specified by the Rare Gas Octet Rule. Thus, in the octahedral molecule SF6, the S +VI cation accepts six donor covalent bond pairs into the empty (3p) and (3d) valence orbitals.
Hydrated Chalcogen Oxides
If the hydrides of Group 6A with the general formula ZH2 are oxidized by oxygen, including OH2 itself, a series of progressively more hydroxylated compounds is produced. These oxidation steps increase the SON of the Group 6A atom in successive steps of +II, eventually initiating the same spontaneous dehydration reactions seen in Group 5A compounds. These reactions are summarized in Figure 10..
|
Oxidation State | |||||||||
| -II | 0 | +II | +IV | +VI | ||||||
| H
y d r a t i o n S t a t e |
0 | ZH2 | <=> | HZOH | <=> | Z(OH)2 | <=> | [Z(OH)4] | <=> | [Z(OH)6] |
| I | I | I | I | |||||||
| -1 |
|
Z | <=> | ZO | <=> | OZ(OH)2 | <=> | O2Z(OH)2 | ||
| I | I | |||||||||
| -2 |
|
ZO2 | <=> | ZO3 | ||||||
Figure 10 Range of Oxidation-Hydration States for Group VI Elements (Z)
Non-existant compounds [ ] are included for completeness
The “nonexistent compounds”, Z(OH)4 and Z(OH)6 cannot actually be studied but their dehydration reactions are included in the reaction scheme to show how the existing compounds can be defined simply as their dehydrated “derivatives”.
The Reaction Mechanism leading to these derivatives, shown in Figure 11, involves the spontaneously release a Hydrogen ion from the fully hydrated molecules by an SN1 Mechanism.
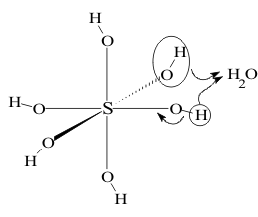
As in earlier Groups, this ion would then react with the (O-H) group beside it in the Structure to form a molecule of H2O which is ejected from the Transition State. This leaves behind a dehydrated product with a new (Z=O) double bond. The increased Polarizing Power on the Z atom then pulls the electron pairs in the remaining (O-H) bonds towards itself making it easier to lose the remaining H atoms as Hydrogen ions, making these compounds successively stronger Bronstead acids. If Z is sulfur or any heavier congener, the sequence starts with the hydride, then leads up the SON sequence to a neutral hydroxyhydride which dehydrates to the Element, then to an unstable dihydroxide, then on to the tetrahydroxide, which dehydrates in two stages to the “ous” acid and the dioxide, then finally to the hexahydroxide, which dehydrates first to the “ic” acid then to the trioxide.
In contrast to the odd SON values of Group 5A atoms, the SONs of Group 6A are even. Consequently, none of these dehydration steps leads to the disproportionation reactions that produce the free radical oxides of nitrogen. However, the decrease in shielding by the core electrons in these Group VI congener cations results in formation of the same type of short double bonds in the oxides of sulfur which were seen in the nitrogen oxides. Thus, instead of having a polymeric Structure similar to P2O3, SO2 is very similar to NO2. Both are gaseous molecular compounds with short (Y=O) and corresponding (Z=O) bonds and together provide an excellent example of the Diagonal Rule of the Periodic Table. These similarities can be observed in the full LCAO MO bond model.
Redox Activities of Chalcogen Elements
The range of SONs available for Group 6A Elements that is shown in Figure 10 means that each Element below oxygen in the Group spans the full range of Reduction-Oxidation Activities. As in Group 5A, the increase in shielding of the valence (np) AOs by the core down this Group increases the Spontaneity of Reduction by ZH2 hydrides, while decreasing the Spontaneity of Oxidation by the oxidizing acids (HO)2ZO2. All of these Activities have been characterized with the compounds of Sulfur and, taking account of these changing shielding effects, the different types of reactions seen for this Element remain similar for the heavier congeners. Typical unbalanced reactions are shown in Table 7.
Table 7 Typical Redox Activities of Group 6A Elements
| Species | SONY | Reduction | Oxidation | Spontaneity | Lability |
| ZH2 | -II | ZH2 + OH -
→
Z + H2O + 2e- |
- | High | Inert |
| ZH(OH) | 0 | Z + 2H2O
→
ZO2 + 4H+ + 4e- |
Z + 2H+ +
2e- →
ZH2 |
Low | Labile |
| Z(OH)2 | +II | - | - | - | - |
| OZ(OH)2 | +IV | ZO32-
+ OH - →
ZO42- + H2O + 2e- |
ZO32-
+ 3H2O + 4e- →
Z + 6 OH - |
Low | Labile |
| O2Z(OH)2 | +VI | - | ZO42-
+ H2O + 2e- →
ZO32- + 2OH - |
High | Inert |
Redox Reaction Mechanisms
As in Group 5A, the complicated changes in Structure that occur during redox reactions of Group 6A atoms are so difficult that they cannot occur in one step but proceed instead as multistep Redox Mechanisms. H2S is a Reducing agent which can saturate (C=C) bonds ;
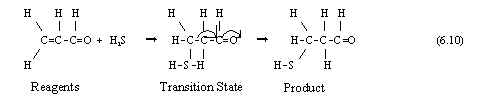
From the same Table, the highest SON found in SO3 makes it an Oxidizing agent, as shown in the Oxidative sulfonation of benzene, which has the stoiciometric form;
H2SO4 + C6H6 → C6H5SO3H + H2 (5a)
but kinetic data show that the acid initially dehydrates in a fast “pre-equilibrium” step ;
(HO)2SO2 Û SO3 + H2O (5b)
to produce the actual oxidizing “reactive intermediate” sulfur trioxide. Then, in the slow rate-determining step, this electron deficient molecule attacks removes a Hydrogen atom from the benzene ring and replaces it a Bimolecular Electrophyllic Substitution, SE2;
SO3 + C6H6 → C6H5SO3H (5c)
to give the final product, benzene sulfonic acid.
Acid-Base Activities of Chalcogen Elements
The wide range of SONs available for Group 6A Elements, also means that each Element after Oxygen spans the full range of Acid-Base Activities. However, the usual increase in shielding of the valence (np) AOs by the core down this Group decreases the Acidity of the hydrated oxides HOZO5. The two reversible Bronstead reactions of Reduced species with any protic solvent, usually water, are ;
ZH2 + H2O ⇌ ZH- + H3O+ (6a)
then with a second Mole of solvent ;
ZH- + H2O ⇌ ZH2- + H3O+ (6b)
The two reversible Bronstead reactions of the Oxidized species with any protic solvent are ;
(HO)2ZO + H2O ⇌ H3O+ + (HO)ZO2-
then with a second Mole of solvent ;
(HO)ZO2- + H2O ⇌ H3O+ + ZO32- (6c)
in which the acid anions are the Spectator Conjugate Bases of each step. All of these Bronstead acid reactions are shown Table 8, with Acid constants where available.
Table 8 Group 6A Acid and Base Strengths
| Species | SONY | Acid Reaction | pKA
( Z = ) |
| ZH2 | -II | ZH2 +
2H2O ⇌
H3O+ + HZ- + H2O ⇌ 2H3O+ + Z2- |
S 7.0,
13.9
Se 3.8, ? Te 2.6, ? |
| (HO)2ZO | +IV | (HO)2ZO +
2H2O ⇌
H3O+ + (HO)ZO2- + H2O ⇌ 2H3O+ + ZO32- |
S 1.89,
7.81
Se 2.62, 8.32 Te 2.48, 7.70 |
| (HO)2ZO2 | +VI | (HO)2ZO2 + 2H2O
⇌
H3O+ + (HO)ZO2- + H2O ⇌ 2H3O+ + ZO42- |
S - 3, 1.96
Se 1.66, ? Te 7.70, 10.95 |
These trends in pKA values again reveal the influence of shielding on the Structures and Activities in a Group of Elements. Going down this Group from Sulfur to Tellurium, the pKA values of the reduced ZH2 acids fall smoothly showing that get stronger, those of the intermediate (HO)2ZO compounds oscillate showing that the acids are similar in strength and those of the fully oxidized (HO)2ZO2 acids rise smoothly showing that these get weaker.
This reversal in Group acidity Activity follows the change in the form of the Structural shielding in the valence orbitals with increasing SON. In the reduced species the shielding becomes less efficient down the Group because the core consists of one, then two (nd)10 shells which shield poorly against the filled ([n+1]p) valence orbital. Thus, the valence electrons, including the (Z-H) bond pairs are pulled progressively into the cores of the heavier atoms, decreasing the (Z-H) bond strength and increasing acid strength.
Conversely, in the oxidized species, the O2- bond pairs are donated directly into all of the empty ([n+1]s,p) valence orbitals, forming the (ns,pσ) and(npπ) bonds of the (Z=O) Structure. The (npπ) bonds form strongly with S +VI at the expense of the strength of the (O-H) σ bonds, making (HO)2SO2 a strong acid. However, in the Se +VI and Te +VI acids, the shape of the one then two (nd)10 shells in the core causes them to shield very strongly against these (npπ) donor bonds. These π bond pairs are pushed back onto the O atoms and strengthen the (O-H) bonds progressively down the Group, reducing the acidities of these species.
Electrochemical Reactivity
In the reactions listed in Tables 7 and 8, the SONs of Group 6A reagents always differ from those of their products by even numbers, 0, +II etc., This indicates that the reagents only differ from the products Structurally by one pair bond. Hammond’s Linear Free Energy Relationship then suggests that reactions involving such minimal Structural changes should only be impeded by minimal Free Energies of Activation. In other words, these reactions will probably follow reversible
Reaction Pathways In all chemical reactions, both the affected valence electron pairs and the atoms of the reagents must rearrange to form the new Structure of the products. In thermal reactions, these two rearrangements occur at the same time in the Transition State. In contrast, in electrochemical reactions, Coulombic forces are applied externally to separate these two processes in both time and space. This separation of processes is achieved in an electrochemical “cell”, shown in Figure 12.
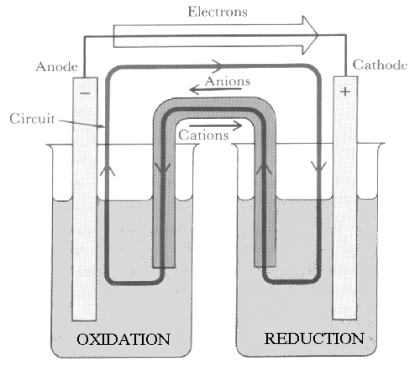
| Electrons are attracted to Positive Anode | Reducing Agents are Oxidized | Chemical Bridge | Oxidizing Agents are Reduced | Electrons are delivered at Negative Cathode |
Figure 12 The Electrochemical Cell and the
Linear Table used as its Schematic Representation
In this cell, the reacting electrons move rapidly from the Anode, where they are produced by reducing reagents in the Oxidation chamber, through a wire outside the reaction medium to the Cathode, where they are consumed by Oxidizing agents in the Reduction chamber. Meanwhile, the positive cations created in the Oxidation chamber can only combine with the anions created in the Reduction chamber much more slowly by passing through the Chemical Bridge inside the reaction medium.
Two types of reaction can occur in these Cells, as defined in Table 9. However, regardless of the Spontaneity of the Reaction, the physical structure of the Cell is always written
Table 9 Electrochemical Cell Properties
| Cell Property | Cell Type | |
| Name of Cell | Electrolytic | Voltaic or Galvanic |
| Electron Flow | Driven | Spontaneous |
| Activity of Cell | Consumes power | Provides power |
in the Standard form given in Figure 12. This maintains a constant direction for the flow of electrons from left to right which corresponds to the flow of the conventional current from right to left. Therefore, to show a reversal of reaction spontaneity and hence of Cell operation, the chemicals in the half-cells are reversed instead of reversing the direction of electron flow.
Within these conventions, the separation of the electron flow from the chemical reagent flow makes it possible to measure the Stoiciometry and the Spontaneity of the reaction separately. The Stoichiometry is defined formally as the number of Moles of electrons exchanged between reagents for complete reaction. It is defined electrochemically as the total charge, C, which must be passed to achieve complete reaction and measured by integrating the current, i, during the reaction. This charge, defined in Coulomb units is converted to Moles of electrons by the Faraday equation, which defines a Mole of electrons as 96,500 Coulombs of electrical charge. This value is called the Faraday constant, F
The Spontaneity of the reaction is defined formally by the sign and magnitude of the Standard Free Energy of Reaction, ΔGoReaction . It is defined electrochemically as the electrical potential of the Cell at the beginning of the reaction and is measured directly between the Anode and Cathode. This potential, defined in volts, is converted to the Standard Free Energy of Reaction by the Gibbs equation which defines a kiloJoule/Mole of electrons as :
ΔGoReaction = n F E0 (7)
where n is the number of Moles of electrons, F is the Faraday constant and E is the cell potential, measured in volts.
To make these concepts practical, the same type of standardization and calibration required for thermodynamic properties is needed to classify these potentials. Again an arbitrary “0” is required for defining E0. This was based on the potential needed to reduce the H + ion to the H atom, represented by the diatomic molecule H2 (gas) at Standard temperature and Pressure ;
2 H + + 2e - ⇒ H2 (gas) (8)
The actual potential of this “Reduction Half Reaction” was then defined arbitrarily to be 0.00 volts. The potentials of all other Reduction Half Reactions were then measured relative to this zero point and tabulated for general use. For practical purposes, more convenient secondary standards are commercially available but “traceable” to the Hydrogen Standards kept in National centres. The full range of these Potentials is shown in Table 10
Table 10 Chemical Range of Standard Reduction Potentials
| Half reaction | Potential | Free Energy | Spontaneous Reaction |
| K+ + e - ® K(s) | -2.95 | Non-spontaneous | oxidation |
| H+ + e - ® ½ H2(g) | 0.00 volts | Standard | - |
| MnO4- + 5 e - ® Mn+II | +1.51 volts | Spontaneous | reduction |
An oxidation Half Reaction is written as the reverse of the Reduction Half Reaction. For example, the Oxidation Half reaction for Potassium (K) becomes ;
K (solid) - e - ® K+ (liq) E o = +2.95V (9)
The Stoiciometry of the full Reaction is then defined as the balanced sum of the Half Reactions but its Spontaneity is defined as the simple sum of the corresponding Reduction and Oxidation potentials;
E0cell = [ E1/2 (red) + E1/2 (oxid) ] (10a)
Since the sign of the Reduction Potential must be reversed to represent the voltage of the Oxidation Half reaction, this equation is usually written with the sign change included ;
E0cell = [ E1/2 (red) - E1/2 (oxid) ] (10b)
so that the voltage in the Reduction Potential table is written in directly without changing the sign.
Electrochemistry on Reaction Pathways
Since the Cell Potential is directly related to the Reaction Free Energy, it follows the Reaction Pathway as the yield goes towards completion. This relationship between the initial and later potentials on the Pathway was put into quantitative form by W. Nernst in 1891, as the equation;
where the second term contributing to Ecell is the voltage generated by change in concentrations of reagents and products along the Pathway. It applies to any type of reversible reaction, Redox or Acid-Base, and is used to describe reaction conditions equally well in batteries or pH metre cells. The essential difference between these two types of reaction can be seen in the ways that the two terms are defined. In a Redox reaction the first term E0cell is defined by equation (10.10b) and the second term by the yield. In an Acid-Base reaction, E0cell is zero because there is no permanent exchange of electrons and the cell voltage depends only on the yield.
Thus, in measurements of the Redox properties of reagents, the value of Ecell is determined at 50% yield when the yield term vanishes exactly. Then Ecell represents only E0cell , which defines the Spontaneity of the reaction under STP conditions. In contrast, in measurements of Acid-Base reaction properties, the value of Ecell represents only the yield. If the yield from exactly 1.0 Moles of initial reagents is measured at equilibrium, the value of Ecell represents a standard yield and defines the equilibrium constants KA for acids or KB for bases, like those given in Table 8 ;