Group 5A The Oxidizing Main Elements
Overall Relationships of Structures to Activities
The Elements of Group 5A again begin in the second row of the Periodic Table because the filling of the (1s)2 orbital of Helium completes the first row of the Table. All Group 5A Elements have electronic configurations [Rare Gas ](ns)2(np)3, with n from 0 to 7
Group 5A Elements; The Pnicogens (“Choking Producers”)
Spatial and Electronic Structures
As in previous Groups, the (ns)2 electrons can penetrate the core of filled p orbitals from all directions but as before, this core provides a shield with no defects which makes this valence pair of electrons unstable. However, this shielding is most effective against (np)2 electrons which can only penetrate to the nucleus from the two ends of its p lobe. Thus, these (np)2 electrons are more easily removed than the (ns)2 electrons. However, in Nitrogen, the [(1s)2] core provides poor shielding and both types of valence electrons can penetrate to the nucleus and the difference in their penetrating powers is small. However the core becomes very effective down the Group to Antimony and Bismuth, so the difference in penetration by valence (ns)2and (np)3 electrons increases, as shown by the decrease in Electronegativity in Table 1.
Table 1 Group V Correlation of Electron Configuration to Electronic Structure
| Element
Name |
Structural
Configuration
[core electrons] (valence electrons) |
r
nm |
η
kJ/M |
χ
kJ/M |
| Nitrogen | [(1s)2] (2s)2 ( 2p)3 | 0.16 | 675 | 725 |
| Phosphorus | [(1s)2(2s)2(2p)6] (3s)2 (3p)3 | 0.19 | 470 | 540 |
| Arsenic | [(1s)2(2s)2(2p)6(3s)2(3p)6] (4s)2 (4p)3 | 0.19 | 440 | 510 |
| Antimony | [(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)10(4p)6](5s)2(5p)3 | 0.22 | 385 | 445 |
| Bismuth | [(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)10(4p)6(5s)2
(4d)10(5p)6] (6s)2 (6p)3 |
0.21 | 365 | 340 |
The net effect of these shielding conditions is that the Effective Nuclear Charge ZEff, begins at moderate values in Nitrogen and decreases down the Group. This leads to a progressive increase in the atomic radius, representing the spatial aspect of atomic Structure of and a decrease in Hardness and Electronegativity.
Bonding Structures
In general, the type of [ M-X ] bond, ionic, donor covalent or covalent, depends on the relative polarizing powers Π and polarizabilities α of the two Elements forming the bond pair. However, in the specific case of Group 5A, these Structural parameters have values intermediate between those found in Elements at the beginnings and ends of Periods. This again makes the Group 5A Elements “electronically amphoteric” so that they usually achieve charge satisfaction by covalent sharing rather than by ionic exchange of electron pairs. In spite of this, it is still very useful to identify the Oxidation Numbers of Group 5A Elements in their compounds in order to describe how electron pairs are rearranged during reactions.
Identifying the SONs
Since these atoms are usually covalently bound within the Structures of compounds, their Oxidation Numbers again cannot be assigned from experimental measurement of the their charges. Instead, as in earlier Groups, the SONs must be assigned theoretically on the basis of the relative Electronegativities of the atoms in the bond, as they were defined in the Rules for Stable Oxidation Numbers in Chapter 2. Following the Octet Rule, the range of Oxidation Numbers is again limited by the Structures of the previous and the next rare gas respectively. Removing all valence electrons to achieve the configuration of the previous Rare Gas gives an upper SON limit of +V Adding enough electrons to fill the valence shell and achieve the configuration of the next Rare Gas gives a lower SON limit of -III for atoms in this Group. Following the Pauli Principle, the intermediate SONs occur when Pauli pairs are preserved. Thus in Group 5A, these are the reduced values of -III, and -I, and the oxidized values of +I, +III, +V.
Electronic Structures of Pnicogen Compounds
When these Rules are applied to Group 5A compounds, they assign SON values over the entire range from the lower to upper Rare Gas limits. Thus, all single bonded compounds have the molecular formulas X3N or X5N. The SON Rules for all of the X3N compounds become;
3(SON)X + 1(SON)N = 0 (1a)
hence, 1(SON)N = 0 - 3(SON)X (1b)
This expression can be used to identify the value of (SON)N in compounds with Elements with Electronegativities lower, similar and lower than Nitrogen, with an χ value of 725kJ/M. The formal SON values of Nitrogen in compounds across this whole range are given in Table 2.
Table 2 SON Values for Nitrogen in X3N Compounds
| Compound | χX | (SON)X | (SON)N |
| Li3N | 290 | +I | -III |
| H3N | 690 | +I | -III |
| NF4 | 1000 | -I | +III |
Reduction to Minimum SONs
In reactions between these Group 5A Elements and less Electronegative metals, Pauling’s Electroneutrality Rule predicts that to minimize the energy of the whole system, the Group 5A atoms would gain electrons into their unfilled (np)3 orbital from the metallic atom. This “reduces” the Group 5A Element to an anion with a minimum SON of -III and “oxidizes” the less Electronegative Element to a cation. For example, in reaction with the Alkali Metals, M, the Group 5A Element, Y would react to form a product ;
3M + Y ⇒ M3Y (2)
Depending on its Polarizing Power, the oxidized species M (+n) may bond to the Group 5A anion, to form either ionic compounds such as lithium nitride, Li3N, or covalent compounds such as ammonia, NH 3 or phosphine, PH3 .
In all of these compounds, the assignment of the minimum SON value of -III to the Group 5A Element defines this Structure as “electron-rich”. This reverses their Structures and Activities from those of the “electron-deficient” Structures of the X3B compounds of Group 3A Elements. In the Structures of the X3B compounds, there were only enough electrons to make the three bonds in and one valence orbital was left unoccupied. Thus, their Activities were dominated by acceptance of electron pairs, reversibly as Lewis acids or at the irreversible reaction limit, as “oxidizing agents”. In contrast, in the Structure of X3Y compounds, the Group 5A atom not only brings enough electrons to make the three (X-Y) bond pairs but also brings an additional lone pair which remains “non-bonding” in the valence shell. In most cases, χY > χX , so that the Activities of these compounds are dominated by donation of this extra electron pair, either as a Lewis base or, in the irreversible reaction limit, as a “reducing agent”.
This donor ability, often called the “Donicity” of the compound, increases down Group 5A because increasing shielding by the core makes the filled (ns)2(np)6valence shells increasingly unstable. The decrease in χ is very large from N to P but from P to As, the shielding by the filled (3d)10 shell is very ineffective and the decrease in χ is minimal. Then, from As to Sb, the addition of the second shield of (4d)10 makes a more effective d-orbital shield and the drop in χ is again large. This alternating pattern of changes in χ down any Group is called “Group Oscillation”.
The “Pnicnine” Compounds
All of the Group 5A Elements form YH3 compounds, which are identified by the suffix “ine” Their Structure and Activity properties are given in Table 3.
Table 3 The Structures and Activities of YH3 Compounds
| Name | Formula | Bond Stability
SB
(kJ/M) |
Bond Type TB ( Δ kJ/M ) | Bond Angle (degrees) | Basicity (kJ/M) |
| Ammonia | NH3 | 391 | 35 | 106.6 | 865 |
| Phosphine | PH3 | 322 | -150 | 93.7 | 770 |
| Arsine | AsH3 | 247 | -180 | 91.8 | - |
| Stibine | SbH3 | 255 | -245 | 91.3 | - |
| Bismuthine | BiH3 | < 200 | -350 | ≈90 |
The changes in Structures and Activities of these compounds follow the changes in Hardness and Electronegativities of the Elements down the Group. Specifically, bond stabilities, SB decrease, bond types, TB, calculated as the difference in Electronegativity between the Element and Hydrogen, reverse from (H+-Y-) in ammonia to (H--Y+) in PH3 and become increasingly hydridic, the (H-Y-H) bond angles decrease and the Bronstead Basicity, measured as the Proton Affinity of these compounds, falls rapidly from NH3 to PH3, then becomes insignificant for the heavier Pnicnines.
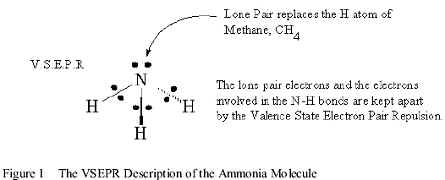
Since these compounds are very important in many applications, many Valence Bond and the Molecular Orbital types of models have been used to describe these Structure and Activity properties. The most commonly used Valence Bond model is Valence State Electron Pair Repulsion theory, while the most familiar Molecular Orbital model is LCAO MO theory. The VSEPR theory describes ammonia as a molecule with three pair bonds and a lone pair. The hybridization of these valence orbitals is defined by distributing all of these valence pairs on the central atom before the interaction with the H atoms is calculated. For these compounds, these pairs occupy an sp3 hybrid valence orbital. If it is assumed that these pairs repel each other equally, this molecule should be tetrahedral, with a (H-Y-H) bond angle of 109.9o, as shown in Figure 1.
The small difference from 106.6o in NH3 is explained by the diffuseness of the lone pair. However, the weaknesses of this model show up in many ways. Experimentally, the rapid decrease in the (H-Y-H) bond angle from ammonia to Bismuthine shows that the assumption of initial sp3 hybridization is completely inappropriate. The bond angle of 90o in Bismuthine implies that the three bonding pairs only occupy the (6p) orbitals of Bi -III. The lone pair has apparently disappeared into the electronic core of the Bismuth atom as a filled (6s)2 orbital because s orbitals penetrate into such cores much better than p orbitals. This progressive change in electronic Structure invalidates the assumption of sp3 hybridization and accounts for the weakening of these bonds, the change in bond type and the reduction in Bronstead basicity.
Theoretically, a simple Coulombic force calculation also shows that this assumption of initial sp3 hybridization is completely inappropriate. In the Structure of NH3, the bond pairs are stable between the N and H atoms because of the Coloumbic attraction force from the positive nuclei on each end of the bond keep the electrons trapped in the middle. For the lone pair however, there is no atom at the outer end, as shown in Figure 2.

Therefore, there is no Coulombic attraction force to hold the electron pair in this hybrid orbital position. Thus any electrons in this position would migrate to the nearest positive charge at the central atom Z.
However, down Group 5A, the VSIP energy separation between the (ns) and (np) orbitals of the Elements, increases, as shown in Figure 3.
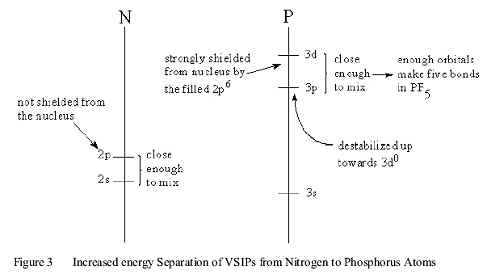
This leads to a decrease in overlap between the (ns) orbital of the central atom and the (1s) orbitals of the H atoms. Thus the (Z-H) bonds become progressively more (Z(np)-H(1s)) in character. Since the angle between the (np) orbitals of the pnicine atom is 90o, the (H-Z-H) bond angle becomes esserntially 90o in the all of the ZH 3 molecules of the heavy Elements of group 5A.as shown in Figure (9.4).
For this reason, the VSEPR model has largely been abandoned in recent molecular models, in favour of the more accurate LCAO models.
The LCAO Description of the Bonding Orbitals in X3Y Molecules
The LCAO MO theory used earlier to describe the bonding in diatomic molecules can be adapted to describe the bonds in these “polyatomic molecules”. The first step is to recognize that all of the H atoms are chemically equivalent in a triangular plane. If their (1s) are all in the same phase, all three orbitals form a Group Orbital with “a1“symmetry. Together, then according to the Woodward-Hoffman criterion, they can then overlap in phase with the Z(ns) orbital to form an a1 or “Totally Symmetric” Linear Combination Molecular Orbital, as shown in Figure 5..
If on the other hand, their (1s) orbitals are of out of phase, they can Linearly Combine into one of two equivalent Asymmetric Group Orbital with “e” symmetry. Together, they can then overlap in phase with one of the Z(np) orbital to form one lobe of an “e” or “Antisymmetric” Linear Combination Molecular Orbital as in Figure 6.
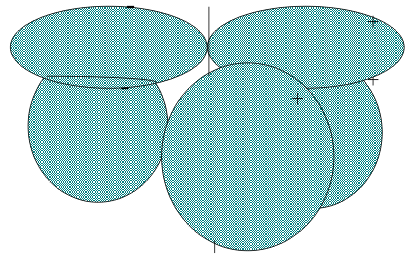
For these in-phase combinations, the Totally Symmetric combination is like a σ bonding MO while the Asymmetric combination is like a π MO. If on the other hand, the relevant Z atom is out of phase with either the a1 or e Group Oritals of 3H(1s), the corresponding a1* or e* antibonding MOs are formed.
Because of their different spatial geometries of a1 and e Group Orbitals, the two types of MO combination achieve two different amounts of overlap . Usually the Totally Symmetric MOs achieve much greater overlaps than the Antisymmetric lobes. Thus, the Totally Symmetric MOs are usually more covalently stabilized than the Antisymmetric MOs and the reverse order must then occur in the corresponding antibonding energies. The Energy Level Diagram for these polyatomic MO energies is constructed the same way as with diatomic MOs and is shown in Figure 7.
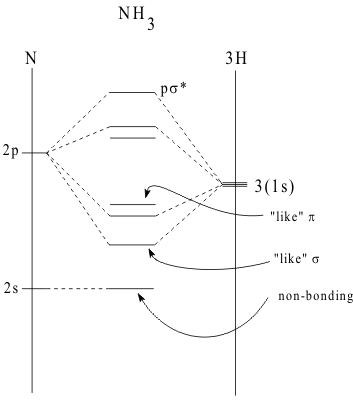
In this diagram, the VSIPs of the Z atom (ns) and (np) orbitals are plotted on the left vertical axis and the VSIP of the H(1s) orbital is plotted on the right vertical axis. The GOs of the three (1s) AOs are indicated by drawing three energy levels together. This occurrence of orbitals with the same energy is called “degeneracy”.
In this case the GOs are labelled as the singly degenerate a 1 and doubly degenerate e GOs. Then, as is done with the diatomic MOs, the energies of the polyatomic MOs are plotted between these vertical axes as the stabilized bonding and destabilized antibonding MOs generated by allowed and forbidden overlap phase conditions respectively. If overlap of the (ns) AO of the Group V atom is ignored, the overlap of the three (np) AOs with the three (1s) GOs of Hydrogen creates three bonding and corresponding antibonding MOs.
By applying the Aufbau Principle, the available valence electrons from the Group V atom and the three H atoms are placed together into the most stable orbitals of the system. Thus, they will first occupy the non-bonding (ns) of the Group V atom then, they will fill the three bonding MOs.
These three bonding MOs made entirely from the three (np) orbitals of the group V atom are all that are needed to make a Stable molecule with the equivalent of one bond to each H atom. The fact that the (ns) orbital is unused and that none of the MOs point at the H atoms but instead one is a σ-like bonding MO pointing straight up the central axis, while the other two are a π-like pair of bonding MOs pointing across the central axis, does not matter to the stability of these molecules. Thus, in the LCAO model, the bond angle in the YH3 compounds is determined by the balance of the overlap in the a1 MO, which pulls the angle inwards against the e overlap pulling the angle open towards a flat triangular shape. As shown in Figure 7, the “lone pair” in the (nsa 1) orbital is not involved in determining the shape of the molecule at all.
Oxidation of the Elements
In contrast, in the reactions of Group 5A Elements and more Electronegative nonmetals, Pauling’s Electroneutrality Rule predicts that, to minimize the energy of the whole system, the Group 5A atoms would lose electrons from either or both of the (ns)2 and (np)3 orbitals to the nonmetallic Element. This “oxidizes” the Group 5A Element to a stable cation of Oxidation Number +III or +V and reduces the nonmetallic Element. However, the Group 5A Elements have high enough intermediate Ionization Potentials that they exhibit stable +III Oxidation Numbers in many compounds. Their Structural parameter values are given in Table 4.
Table 4 Electronic Structures of Stable Group V Oxidation Number +III
| Stable
ON |
Configuration
[Core](valence) |
r
nm |
η
kJ/M |
(106)α
(nm3)M/kJ |
χ
kJ/M |
Π (10-4)
kJ/Mnm |
| N +III | [ He ](2s)2(2p)0 | ≈0.04 | 1450 | 0.04 | 6025 | 15.1 |
| P +III | [ Ne ](3s)2(3p)0 | ≈0.06 | 1025 | 0.2 | 3925 | 6.6 |
| As +III | [ Ar ](4s)2(4p)0 | ≈0.07 | 1085 | 0.35 | 3800 | 5.4 |
| Sb +III | [ Kr ](5s)2(5p)0 | 0.092 | 920 | 0.8 | 3350 | 3.6 |
| Bi +III | [ Xe ](6s)2(6p)0 | 0.11 | 960 | 1.4 | 3425 | 3.1 |
In a few other compounds, the most oxidizing of the non-metal Elelents are strong enough to oxidize these Group 5A Elements to the maximum SON, +V. In this form, the Group 5A Elements have properties given in Table 5.
Table 5 Electronic Structures of Stable Group V Oxidation Number +V
| Stable
ON |
Configuration
[Core](valence) |
r
nm |
η
kJ/M |
(106)α
(nm3)M/kJ |
χ
kJ/M |
Π (10-4)
kJ/Mnm |
| N +V | [ He ](2s)0 (2p)0 | 0.005 | 21900 | 6x10-6 | 31300 | 625 |
| P +V | [ Ne ](3s)0 (3p)0 | 0.02 | 7500 | 0.001 | 13700 | 68.5 |
| As +V | [ Ar ](4s)0 (4p)0 | 0.035 | 3150 | 0.013 | 8700 | 9 |
| Sb +V | [ Kr ](5s)0 (5p)0 | 0.06 | 2500 | 0.085 | 7900 | 4.2 |
| Bi +V | [ Xe ](6s)0 (6p)0 | ? | 1550 | ? | 6950 | ? |
The Group 5A Halides
As with Group 4A Elements, the simplest oxidation reactions occur when the nonmetallic Element acquires the valence electrons of the Group 5A atom, becoming “reduced” to a stable anion, while the Group 5A atom is oxidized to a cation. In reaction with the Halogens, X2 ;
2M + 3X2 ⇒ 2MX3 (2)
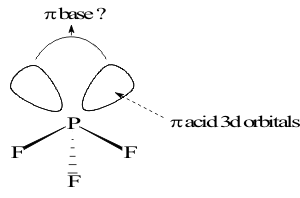
and the resulting anion can then form single σ bonds to the Group V cation. However, the Polarizing Power of Group 5A cations is so high that the Halide ions are forced to provide donor covalent bonds to the oxidized species which results in formation of molecular compounds instead of salts. In general, these compounds have formulas X = YX3 or YX5 corresponding to their SONs. However, like the hydrogen compounds, these halide derivatives are electron-rich. As in that case, the poor shielding in N +III and +V allows the (2s) and (2p) valence orbitals to overlap with the a1 GO but the much higher shielding in the heavier congeners.
This attracts the non-bonding pair of valence electrons into the (ns) orbital, leaving the (Y-X) bond pairs essentially in the three (np)x,y,z lobes, with their (X-Y-X) bond angles of 90o. However, this energy separation of (ns) and (np) valence orbitals brings the (np) close in energy to the empty (nd) orbitals of these heavier cations. This is especially true when the Electronegativity of X is high, such as with F in PF3 or PF5(g) . The Stability of these empty 3d orbitals and allows them to accept one or two extra pairs of bonding or non-bonding electrons, beyond the number specified by the Rare Gas Octet Rule. This is called an “Expand Valence” and is found in heavy cations from Group 5A to Group 8A. It makes all of these cations electron-deficient giving them Lewis acid character. They can bond to electron rich species by accepting π electron pairs into their empty (ndπ) MOs, shown in Figure 8.
Hydrated Group 5A Oxides
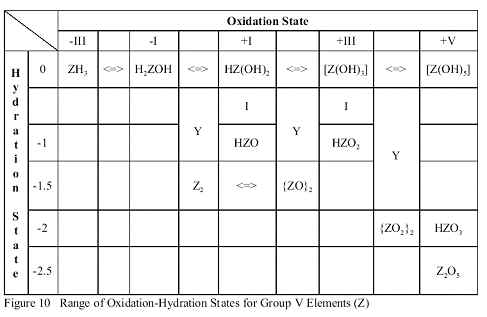
If the hydrides of Group 5A, starting with NH3 are oxidized by oxygen itself, a series of progressively more hydroxylated compounds is produced, as shown in Figure 9.
| SON | -III | -I | +I | +III | +V |
| Initial Formula | H3N | H2NOH | HN(OH)2 | N(OH)3 | N(OH)5 |
| Final Formula | H3N | H2NOH | HNO | HNO2 | HNO3 |
| Compound Name | Ammonia | Hyroxylamine | ? | Nitrous Acid | Nitric Acid |
Figure 9 The range of SONs available for Group 5A Elements, as shown for Nitrogen,
This oxidation increases the SON of the Group 5A atom in successive steps of +II, which eventually increases its Electronegativity above that for Hydrogen. Then, one or more of the (O-H) bond pairs is attracted to the (N-O) bond, which releases a Hydrogen ion by an SN1 Mechanism. This ion reacts with the (O-H) group beside it in the Structure to form a molecule of H2O which is ejected from the Transition State. This leaves behind a dehydrated product with a new (N=O) double bond, as shown in the sets of reactions on successively oxidized derivatives of ammonia in Figure 10.
 |
The increased Polarizing Power on the nitrogen then pulls the electron pairs in the remaining (O-H) bonds towards itself making it easier to lose the remaining H atoms as Hydrogen ions. Thus, these compounds become successively weaker Bronstead bases, from ammonia to hydroxylamine hydroxide, up to the neutral but unstable [HNO], then they become successively stronger Bronstead acids through the weakly acidic nitrous acid, HONO, up to the very strong nitric acid, HONO2.
As the Bronstead acidity of these compounds increases, so does their tendency to dehydrate a second time. However, because the SON values of the nitrogen atom are always odd these compounds have only one Hydrogen left in their Structures. Thus, unlike the first Unimolecular dehydration, this second dehydration has to proceed by a Bimolecular Reaction Mechanism in order to unite the two Hydrogen atoms required to form one water molecule. Even then, the reaction cannot be balanced for the oxygen atoms with two molecules of the same nitrogen SON. The dehydration can only proceed if two molecules of different nitrogen SON are involved. Then, as the water is ejected, the nitrogen atoms “split their SON difference” forming two Moles of the appropriate “anhydrous” oxide of nitrogen, either nitrous oxide, NO, or nitric oxide, NO2. Since the SON of nitrogen must now be an even value, +II or +IV, their electronic Structure must involve an unpaired valence electron. Such molecules are called “free radicals” and are usually extremely reactive since they rapidly reform pairs of electrons wherever possible. However, the nitrogen oxides are kinetically Inert and persist in the atmosphere as the well known “NOX” gasses of air pollution. In order to explain this unexpected Inertness of ·NO, the complete LCAO MO description of its diatomic electronic Structure, as shown in Figure 11.
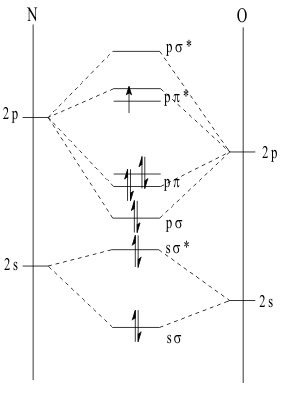
. It closely resembles the Structure of CO but the Aufbau principle places the last unpaired electron in the pp* orbital, giving the free radical Structure. The Bond Order is determined as ;
(2sσ)2 + (2pσ)2 + 2(2pπ)2 ] - [ (2sσ*)2 + (2pπ*)1 ] = 2 ½ (3)
While these free radical oxide nitrogen molecules are unusually unreactive, they are not completely Inert. the pure gases will couple their antibonding (2pπ*)1 electrons to make a weak π bond in a dimer molecule, as shown in Figure 10. However, since the bond is very weak, this dimerization Reaction Pathway is reversible and the process reaches an equilibrium before completion.
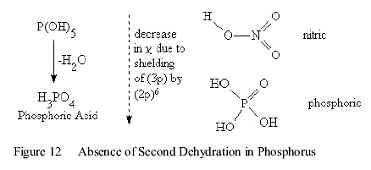
Oxidation of Congeners
The oxidation-dehydration reactions of the heavier congeners, of Group V, Phosphorus and Arsenic follow the same initial steps taken by the Nitrogen compounds but do not follow the second dehydration step to form discrete molecules by disproportionation reactions shown in Figure 9. This difference in Activity is entirely due to the increase in shielding by the core electrons in these heavier congeners. This reduces the Stabilities of the (np) valence orbitals, which increases the orbital radius. In turn, this increases the length of the (P-O) bond, as shown in Figure 13.
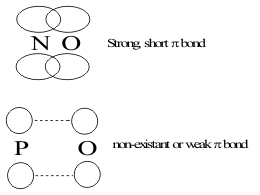
to the point that pπ overlap is lost and eliminates the formation
of the (npπ) bonds required to stabilize the unpaired electron in the (npπ*)
antibonding MO. Thus, the second step of dehydration becomes much more difficult
because there is no stable ·PO free radical product.
Instead, the second dehydration step leads to formation of the singly-bonded oxide polymers of P +III and P +V shown in Figure 14
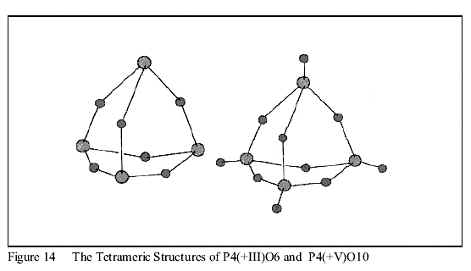
Arsenic (As) behaves the same way. Thus, in both cases, the SON assignments of the Element within the compound alone only give a qualitative explanation the Structure and Activities of these oxides and, by themselves, cannot predict the Structures, the spontaneous disproportionations or the polymerizations of the oxidized derivatives. These Activities can only be described or predicted from the full LCAO MO model.
Redox Activities of Pnicnogen Elements
The range of SONs available for Group 5A Elements, as shown for Nitrogen in Figure (6.9), means that each Element spans the full range of Reduction-Oxidation Activities. However, the usual increase in shielding of the valence (np) AOs by the core down this Group increases the Spontaneity of Reduction by YH3 hydrides, while decreasing the Spontaneity of Oxidation by the oxidizing acids HOYO5. All of these Activities have been characterized with the compounds of Nitrogen and, taking account of these changing shielding effects, the different types of reactions seen for this Element remain similar for the heavier congeners. Typical unbalanced reactions are shown in Table 6
Table 6 Typical Redox Activities of Group 5A Elements
| Species | SONY | Reduction | Oxidation | Spontaneity | Lability |
| YH 3 | -III | YH3 + O2 →
YO + H2O |
- | High | Inert |
| H2Y(OH) | -I | YH3OH + Fe3+ →
Y2O + Fe2+ |
? | Low | Labile |
| HOYO | +III | HOYO +
H+ + I- →
YO + H2O + ½ I2 |
HOYO +
xH2O →
YO3- + 3H+ + e- |
Low | Labile |
| HOYO2 | +V | - | HOYO2 + C6H6 →
C6H5YO2 + H2 |
High | Inert |
As should be expected in this series, the Spontaneity of Reduction decreases and the Spontaneity of Oxidation increases as the SON increases. However, the Labilities of both types of Reaction show the same trends. In other words, the size of the Free Energy of Activation seems to depend directly rather than inversely on the size of the Free Energy of Reaction. The result is a very odd paradox, “the more a reaction should happen the less likely it will happen”.
This phenomenon has been noticed in most of the Inert Reactions of p-block Elements and was first called the “Linear Free Energy Relationship” by George Hammond in 1955. He postulated that whenever there was a big change in the Free Energy of Reaction there had to be a big change in Structure from Reagents and Products, requiring a large Free Energy of Activation. This resolves the paradox by showing that the actual linear relationship between the Free Energies of Reaction and Activation is exactly opposite to the one expected on simple physical assumptions.
Since the QSARs for different types of compounds often show this type of “anti-linear” or curved form, chemistry itself is usually referred to as a “non-linear science”. This description of chemistry emphasizes the fact that none of the Activities of compounds can be predicted as simple sums of the Activities of their atoms.
Redox Reaction Mechanisms
These complicated changes in Structure which illustrate this anti-linearity of the LFER are often so difficult that they cannot occur in one step. Instead they require multistep Mechanisms of Redox Reaction. The oxidative nitration of benzene, Table 6, has the stoiciometric form;
HNO3 + C6H6 → C6H5NO2 + H2 (4a)
but kinetic data show that the acid initially “predissociates” in a fast “pre-equilibrium” step;
2HONO2 Û NO2+ + NO3- + H2O (4b)
to produce the actual oxidizing “reactive intermediate” nitronium ion NO2+. Then, in the slow rate-determining step, this ion attacks benzene in a Bimolecular Electrophyllic Substitution, SE2;
NO2+ + C6H6 → C6H5NO2 + H+ (4c)
to give the final product, nitrobenzene.
This anti-linear LFER explains why it is possible to synthesize and retain compounds with high Spontaneity for decomposition. All the chemistry of Life is based on this anti-linearity as is the “Stability” of most industrial compounds, especially fertilisers and explosives. The simple salt NH4NO3 which is often used for both purposes provides a clear example. Hess’ Law ;
ΔGReaction = ΣΔG°Product - ΣΔG°Reagent (5)
shows that, in the presence of acid, the reaction ;
NH4NO3 + 2H+ → N2 + 3H2O (6a)
has a strongly negative Free Energy of Reaction ;
ΔGReaction = {ΔG(N2) + 3ΔG(H2O)} - {ΔG(NH4NO3) + 2ΔG(H+)} (6b)
= { 0 + 3(-242)} - {(-365) + 2(-92)} = - 177 kJ/M (6c)
and that the solid salt should decompose explosively from to gaseous products. The fact that this does not happen at STP is entirely due to a very large positive Free Energy of Activation barrier which arises because this is a multi-step process in the solid state.
Acid-Base Activities of Pnicnogen Elements
The range of SONs available for Group 5A Elements, as shown for Nitrogen in Figure 9, also means that each Element spans the full range of Acid-Base Activities. However, the usual increase in shielding of the valence (np) AOs by the core down this Group increases the Basicity of the YH3 hydrides, while decreasing the Acidity of the hydrated oxides HOYO2. Taking account of these changing shielding effects, the different types of reactions seen for this Element remain similar for the heavier congeners. Typical Bronstead reactions and conditions are shown in Table .8. The reversible Bronstead reaction of Reduced species with any protic solvent, usually water, is;
YR3 + H3O + ⇌ YR3H+ + H2O (7a)
Base (Lewis donor) Acid (Lewis acceptor) Complex Spectator
where R may be any substituent on Y. and the uncomplexed solvent is a Spectator Conjugate Base to the reaction. The reversible Bronstead reaction of Oxidized species with any protic solvent is ;
YR (OH) + H2O ⇌ YR (O) - + H3O + (7b)
Acid (Lewis acceptor) Base (Lewis donor) Spectator Complex
where again R may be any substituent on Y and the acid anion is the Spectator Conjugate Base. These classes are summarized in Table 7, with the relevant acid and Base constants, when available.
Table 7 Relative Acid and Base Strength
| Species | SONY | Base Reaction | Acid Reaction | pKB
( Y = ) |
pKA
( Y = ) |
| YH 3 | -III | YH 3 +
H2O ⇌
YH4OH |
- | N
4.75
P (DCP) |
|
| H2Y(OH) | -I | YH3OH +
H2O ⇌
YH4(OH)2 |
- | N
8.2
P (DCP) |
|
| HOYO
[(HO)2YO] |
+III | - | HOYO + H2O
⇌
H3O+ + OYO- |
→ | N
3.15
P 2.0 As 9.3 |
| HOYO3
[(HO)2YO3] |
+V | - | HOYO3 +
H2O ⇌
H3O+ + OYO2- |
→ | N <
1
P 2.15 As 2.24 |
The two forms of the Oxidized species represent the absence of the second dehydration step beyond Nitrogen. (DCP) means that the compound decomposes by Redox reactions in protic solvents.