Group 3A; The Acceptor Covalent Main Elements
Overall Relationships of Structure to Behaviour
The Elements of Group 3A also begin in the second row of the Periodic Table because the filling of the (1s)2 orbital of Helium completes the first row of the Table. However, they only appear, in the long form of the Periodic Table after the gap created by the Transition Elements in Row 4, as shown in Figure 1;.
All Group 3A Elements have the electronic configuration [Rare Gas ](ns)2(np)1, where again, n has values from 2 to 7. The correlation of the configurations to atomic Structure is shown in Table 1
.
Table 1 Group 3A Correlation of Electron Configuration to Electronic Structure
| Element
Name |
Structural
Configuration
[core electrons] (valence electrons) |
r
nm |
η
kJ/M |
χ
kJ/M |
| Boron | [(1s)2] (2s)2(2p)1 | ≈0.09 | 385 | 420 |
| Aluminum | [(1s)2(2s)2(2p)6] (3s)2(3p)1 | 0.14 | 265 | 315 |
| Gallium | [(1s)2(2s)2(2p)6(3s)2(3p)6] (4s)2(3d)10(4p)1 | 0.15 | 280 | 300 |
| Indium | [(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)10(4p)6] (5s)2 (4d)10(5p)1 | 0.17 | 310 | 250 |
| Thallium | [(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)10(4p)6(5s)2
(4d)10(5p)6] (6s)2 (4f)14(5d)10(6p)1 |
0.17 | 235 | 350 |
As in Groups 1A and 2A, the (ns)2 electrons can penetrate the core of filled p orbitals from all directions but as before, this core provides a shield with no defects which makes this valence pair of electrons unstable. However, this shielding is even more effective against the (np)1, electron which can only penetrate to the nucleus from the two ends of its p lobe. As a result, this (np)1 electron is more easily lost than (ns)2 electrons. Since the [(1s)2] core provides poor shielding in Boron, both types of valence electrons can penetrate through to the nucleus quite well and this difference in their penetrating powers is not important. However the core becomes very effective down the Group to Indium and Thallium, so the difference in penetration by (ns)2 and (np)1 electrons becomes much bigger. The result is again a progressive increase in the atomic radius, representing the spatial aspect of atomic Structure and progressive decreases in Hardness and Electronegativity, representing the electronic aspects of atomic structure from Boron down to Thallium, Table (7.1). As in Groups 1A and 2A, the Hardness and Electronegativity of Group 3A Elements begin at moderate values in Boron and decrease down the Group to Thallium
Oxidized Ions of Group 3A Elements
From the electronic Structure of Group 3A Elements, both Stable Oxidation Numbers +I and +III are predicted from the stepwise loss of the three valence electrons, first of the only p electron, then of the two s electrons. The loss of further electrons in chemical structures is limited by the core of filled orbitals of the previous principal quantum number. These differences in penetration mean that a separate Stable Oxidation Number of +I can only be achieved if the shielding of the p electron is significantly more effective than the shielding against the s electrons. Since all shielding in Boron is very poor, no distinction in shielding against p or s electrons is possible and it can only be oxidized to the one stable Oxidation Number +III. However, the shielding in Indium and Thallium is very strong and the difference in shielding against p as opposed to s electrons now becomes important. These two Elements can therefore be oxidized easily to +I, losing only the poorly penetrating (np)1 electron and only to +III with more difficulty. Using the definitions in equations (R2.1) and (R2.2), the values of all these Structural properties for the stable Oxidation Number +III of this Group are given in Table 2.
Table 2 Electronic Structures of +III Oxidation Numbers of Group 3A
| SON | Configuration
[Core](valence) |
r
nm |
η
kJ/M |
(106)α
(nm3)M/kJ |
χ
kJ/M |
Π (10-4)
kJ/Mnm |
| B +III | [ He ](2s)0(2p)0 | 0.012 | 10600 | 0.0005 | 14500 | 120 |
| Al +III | [ Ne ](3s)0(3p)0 | 0.053 | 4400 | 0.1 | 7100 | 13.5 |
| Ga +III | [ Ar ](4s)0(4p)0 | 0.062 | 1600 | 0.45 | 4500 | 7.3 |
| In +III | [ Kr ](5s)0(5p)0 | 0.08 | 1250 | 1.23 | 4000 | 1.6 |
| Tl +III | [ Xe ](6s)0 (6p)0 | 0.088 | 500 | 4.1 | 2500 | 0.6 |
The trends in the Periodic Table again indicate that, except for Boron, the Hardness and the Electronegativity of most Group 3A Elements is less than that of most nonmetallic Elements. Thus, when the metallic Group III Elements, Al to Tl, come into contact with most non-metals, Pauling’s Electroneutrality Rule again predicts that, to minimize the energy of the whole system, the Group III Elements will lose one or all three of their valence (ns)2(np)1 electrons to the non-metallic Element. This again oxidizes the Group 3A Element to a stable cation, with Stable Oxidation Number +I or +III. At the same time, the nonmetallic atom acquiring these valence electrons from the Group 3A atom is “reduced” to a negatively charged anion. This new anion can then form a new bond with the Group 3A cation. For example, in contact with Halogen molecules, X2, Group 3A Elements react to form donor-covalent “trihalide” molecular compounds;
2M + 3X2 ⇒ 2M(+III)X(-I)3 (1)
With other anions, any type of chemical bond may be formed to the Group 3A cation.
Reduced Group 3A Elements
An analogous limit to chemically available Stable Oxidation Numbers occurs when attempts are made to add electrons to the neutral Group 3A atoms in chemical bonds. The valence p orbital has 5 unoccupied electron positions. Since electrons in a common orbital are all at the same average radius, they do not shield each other effectively from the nucleus, so the Electron Affinity of this unfilled p orbital is significant. This defines the upper chemical limit of electron occupancy as the Structure of the next Rare Gas and the maximum negative Oxidation Number as -V. In practice not even Boron has a high enough Electron Affinity to achieve this Stable Oxidation Number in known chemical compounds but it routinely achieves B -III in its “borides” with an electron configuration of up to (ns)2(np)4. By acquiring these 3 valence electrons, the boron oxidizes the associated metallic atom to a positive Stable Oxidation Number. Since the degree of reduction of Boron is variable, there are many types of boride compounds, including those with the formulas;
(Metal-rich) M4B ⇒ MB ⇒ MB12 ⇒ MB66 (Boron-rich) (2)
In all these cases, the boride anions can form any type of chemical bond between themselves and to the metallic cations.
B +III Compound Structures
The Polarizing Power of Boron, with a Mulliken value of 120kJ/M/nm, is just high enough to make it the first “nonmetallic” Element in the Periodic Table, apart from Hydrogen. The main consequence of this is that it forms “heteronuclear covalent bonds”, (A-B), in compounds with all the other nonmetallic Elements. However, since the Electronegativity of Boron is always lower than that of the other nonmetallic Element in these bonds, Boron always behaves as the positively charged partner in the bond and is always assigned the positive Stable Oxidation Number +III.
The Boron Hydrides
From previous Sections, the Polarizing Powers of B and H are;
Π (Be +III) [120 kJ/M/nm] < Π (H +I) [200kJ/M/nm] (3 )
and the heteronuclear covalent compound formed between Boron and Hydrogen requires electron transfer from Boron to Hydrogen. Therefore, the Boron is theoretically oxidized to +III and the Hydrogen reduced to -I. The final compound is defined as Borane, with the chemical formula ;
B (+III)H(-I)3
but due to the theoretical charge distribution it is sometimes named “Boron Hydride”, as if it were a fully ionic salt.
While this Structure satisfies the formal charge requirements of both the B +III and H -I species in these bonds, BH3 does not actually exist as an isolated, covalent molecule. To satisfy these formal charge requirements, each Hydride -I species donates an electron pair from its filled HOMO, (1s)2 into the LUMOs, (2s)0(2p)0 of the B +III species. This set of 6 electrons, however, does not fill the (2s)0(2p)0 “shell”, which would actually require 8 electrons. As noted before, electrons in equivalent orbitals all have the same average radius and do not shield each other from the nucleus. Thus, the two unfilled positions in the (2s)0(2p)0 shell of B +III offer stability equal to that of the already filled lobes of the shell. Consequently, these two “holes” in the (2s)0(2p)0 shell have high Electron Affinity and BH3 is therefore described as an “electron deficient”compound. Since filling all eight holes in the (2s)2(2p)6 shell mimics the structure of the next Rare Gas, Neon, this tendency to fill the shell is called the “Octet Rule” for stable structures.
Since no single borane molecule can complete its electronic octet internally, its electron deficiency forces it to act as a Lewis acid and seek the required electron pair from any external Lewis base. In pure gaseous borane however, every electron deficient molecule would act as a Lewis acid. The only source of electron pairs is in the (B-H) bonds of any other borane molecule. Even then, this pair is only available on a shared basis, so the most stable structure is reached by the Symmetric sharing of two bond pairs by two molecules, as shown in Figure 2
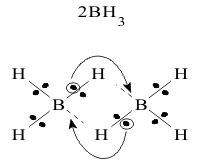
This stable form of borane is therefore called “diborane” and the new shared bonds, with only one electron pair spanning over a B-H-B “bridge” is often called a “three-centre” bond. This process of making three centre bonds can continue on the two other unshared B-H bonds on B atom, to form much higher “polymers” including B4H10 and B5H11. They are all nonpolar covalent molecules, with characteristic low melting and boiling points and low solubility in polar solvents like water.
This process of polymeric bond formation can continue to build a 3D structure ending in the structure of boron as the Element in a spherical B12 molecule. This bonding of B to itself is called “catenation”. This is characteristic of some nonmetallic Elements with Polarizing Power restricted a narrow range of Π values, 120 to 180 kJ/M/nm, found especially for B, C, and N.
The Boron Halides
Similar bonding occurs between B +III and other “univalent” nonmetallic Elements, especially F -I or Cl -I . These have the same Structure as borane, making the neutral compounds BX3, which are again electron deficient because their electronic Structures do not satisfy the octet rule. They are again Lewis acids, seeking the “missing” electron pair. However, the equivalent dimer to diborane, with a formula of B2F6, does not form because F is the most Polarizing Element in the Periodic Table and does not donate a second pair to make a bridge. So the only remaining option for BF3 is to act as a simple Lewis acid and accept polar covalent donor pairs from any Lewis base to form a Lewis complex product as shown in Figure 3..
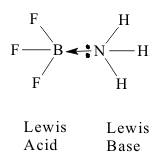
The Boron Oxides and Bronstead Amphoterism
Boron can equally react with “divalent” nonmetallic Elements, like oxygen to form the “stoiciometric” “binary” products like boric oxide ;
B3+ + O2- ® B2O3 (4)
Since the Polarizing Power of oxygen is less that of fluorine, it donates extra bonding pairs to the electron deficient B +III and forms bridge bonds between B3+ ions. In fact bridge bonding is so strong that the molecules polymerize completely to a solid Structure, without any intermediate small molecules.
The oxide ions have such an excess of electron density that even after satisfying the charge requirements of all the B +III atoms, they can still donate charge to other Lewis acids. This makes boric oxide reactive towards all Bronstead Acids which supply H +I ions. Thus ;
B2O3 + 3H2O ⇒ 2 B(OH)3 (5)
However, the close similarity of the Polarizing Powers of B +III and H +I means that the (B-O) and (O-H) bonds are equally strong. Thus, when this “hydrated oxide” B(OH)3 is places in a protic solvent, it can respond to high or low pH conditions by acting as a Bronstead acid or base.
B +III Compound Activities
The quantity of an acid of any strength in solution can be estimated by titration against a calibrated strong base. However, the progress of the titration procedure depends strongly on the strength of the acid being assessed. To illustrate the progress of a titration procedure in detail, the estimation of the concentration of B(OH)3 in water provides a typical example. These estimations are calculated with three different approximations, in the three distinct “Regions” of the Titration
Curve, shown in Figure 4.
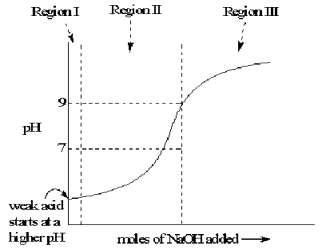
Region I; Before titration begins, the hydrated oxide may react with water as a Bronstead acid:
B(OH)3 + H2O Û BO(OH)2- + H3O+ (6a)
and its equilibrium condition can be quantitated by its acid constant in the usual way to give ;
This very small equilibrium constant shows that the hydrated oxide only dissociates slightly in water and acts as a very weak acid. Therefore, the pH of a 0.10M solution of this compound in water can be calculated using the approximate concentrations of reagents and products of the reaction;
[B(OH)3] [H2O] Û [BO(OH)2-] [H3O+]
before equilibrium: 0.10 1.0 0 0
after equilibrium: 0.10-x 1.0 x x
but, since x < 0.1 M, these concentrations may be substituted into the expression for KA (6b) ;
![]()
Then, by rearranging this equation algebraically to isolate the unknown x
and by taking the negative log of this value, the pH of the initial solution can be determined ;
Region II ; When titration begins, in Region II of the procedure, calibrated quantities of a strong base are progressively added to convert the weak acid irreversibly to the corresponding salt. This follows a reaction equation symbolized by the one-way arrow, ⇒ ;
B(OH)3 + NaOH ⇒ [BO(OH)2 ] - + Na+ + H2O (7a)
Thus, at any point during the titration procedure, after a quantity Z of acid has been converted to the product, significant amounts of both the reagent B(OH)3 and the product [BO(OH)2 ] - are present together in the reaction solution. If the titration is stopped at any point, the remaining, untitrated acid still dissociates, releasing H3O+ according to equation (4.11a), and therefore continues to control the pH of the solution. The resulting pH is calculates as in Region I but takes account of the reduced concentration of the remaining acid in the equilibrium at this point. Thus the calculation uses the adjusted approximate concentrations of reagent and product as follows ;
[B(OH)3] [H2O] Û [BO(OH)2-] [H3O+]
before equilibrium: (0.10 - Z) 1.0 Z 0
after equilibrium: (0.10 - Z - y) 1.0 ( Z - y ) y
![]()
but, since y < 0.1 M,
By algebraic rearrangement, the unknown can be isolated fro for any point in Region II;
This approximate calculation of the unknown, representing [H3O+ ], can be generalized to ;
![]()
and rearranged to isolate the unknown concentration of [H3O+ ] at any point during the titration ;
then by taking negative logs of both sides of this equation ;
![]()
Therefore:
![]()
or in the general Bronstead definitions:
![]()
This equation. (7g), is known as the “Henderson-Hasselbalch” equation and is used for defining the pH inn any part of in Region II and coincidentally for any Bronstead acid-base “buffer”.
Region III: When titration is completed, in Region III of the procedure, a sufficient quantity of the calibrated strong base has been added to react all of the initial acid to its salt. This is called the “equivalence point” of the titration, because the amount of added strong base is now equivalent to the amount of original acid. At this point the only species in solution should be the anion of the acid. However, weak acids are poorly dissociated and their anions tend to reacquire their lost Hydrogen ions from any available source. Since the protic s0lvent water spontaneously dissociates to produce a small concentration of Hydronium ion, these weak acid anions reassociate with these released Hydronium ions to remake some undissociated acid even at the equivalence point. Since these Hydronium ions came from water, this reassociation reaction is called the “hydrolysis” reaction of the anion ;
[BO(OH)2]- + H2O Û B(OH)3 + OH- (8a)
This process can again be quantitated by its equilibrium constant in the usual form ;
![]()
and the pH of the equivalence point solution in Region III of any titration can be calculated as ;
[BO(OH)2-] [H2O] Û [B(OH)3] [OH-]
before equilibrium: 0.10 1.0 0 0
after equilibrium: 0.10-y 1.0 y y
![]()
This is very similar to the calculation of pH in Region I, except that this is a back reaction in which;
![]()
which can be simplified by recognizing that the definitions of both the acid and water equilibrium constants appear when the constant is expanded with [H2O] in the numerator and denominator:
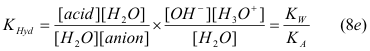
and hence it is not necessary to define a separate concept or calculation for KHyd These Regions are summarized in Table 3
Table 3 Approximation Regions for pH in Titrations
| Region | Chemical System | Approximation for pH Calculation |
| I | pure acid | pKA |
| II | acid-salt mixture | Henderson-Hasselbalch |
| III | pure salt | pKHyd |
Bronstead Buffer Activity
From Figure 4, the pH in Region II changes very slowly, while in Region III it changes very fast. Any system which resists change effectively, like the borate ion in its Region II, is called a “buffer”. This preferential buffering action of Region II comes from the difference between the chemical systems in these two Regions, a mixture in Region II verus a single compound in Region III. In this case the buffering mixture is able to keep the solution near a constant pH value during most of the titration, minimizing the change imposed by addition of base to the solution. This Activity is a direct application of “Le Chatelier’s principle” that any system will respond as well as it can to minimize the effects of a change in conditions.
From this it is clear that buffer solutions have two completely separate properties.
1 The pH of the system is defined in the Henderson-Hasselbalch equation, (7.7e) by two terms. The strength of the acid, defined by pKA, sets the mid-point of the buffering pH range and the deviation from this value is defined by the log of the ratio of conjugate acid and base.
2 The “buffering capacity” of the system is defined by the actual concentrations of conjugate acid and base. At any pH defined by the concentration ratio, the actual buffering capacity depends directly on the quantity of acid (base) available to be consumed by any added base (acid).
Bronstead Polyacid Activity
The stoichiometric formula of hydrated boric oxide, B(OH) 3 suggests that it could have up to three dissociable Hydrogen ions in its Structure. This is actually the case, and titration with strong base forms the salt sodium borate, Na3BO3 ;
B(OH)3 + OH- ⇒ [B(OH)2(O)]- + OH- ⇒ [B(OH)(OH)2]2- + OH-⇒ Na3BO3 (9)
Because they have more than one dissociable Hydrogen ion, compounds like B(OH) 3 are called “polyprotic acids”. However, these Hydrogen ions are not all lost at once at a fixed pH during titration. Instead they are removed by strong base one at a time at very different pH values. This results in a characteristic step pattern in the titration curve, shown in Figure 5, with a separate equivalence point distinguishable for the equivalence point of each dissociable hydrogen ion.
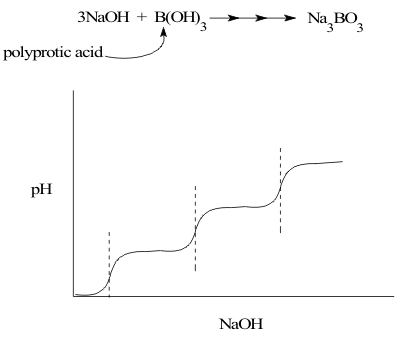
The reason for this “polyacid” behaviour is simple. As each Hydrogen ion is removed, it leaves behind a negative charge on the remaining, anionic boron species. This strengthens the bond to any remaining positively charged Hydrogen ions and makes it more difficult to remove them with base.
The same process occurs during dehydration with heat. Each B(OH)3 unit can lose 1.5 Moles of H2O, so there is a stepwise loss of water during regeneration the original, dry oxide, B2O3.
In both cases, the system of [B-O-H] single bonds is replaced by new [B=O] double bonds. These bonds are formed by electron pair donation from the filled (pπ) orbitals of the electron-rich oxide ions to the empty (pπ) orbitals of the electron- deficient Boron atom. This Lewis π complex formation is common in the oxygen compounds of all high SON forms of Main Block Elements
Diagonal Rule Description of Group 3A Structures
The Sequences of Elements that define the Diagonals of the Periodic Table display many common Structures and Activities. These empirical correlations are known as the Diagonal Rules. The theoretical basis of these empirical correlations can be discovered by looking at the constancy of the purely electronic Structural parameters, Hardness η and Electronegativity χ and the spatio-electronic Structural parameters, polarizability α and polarizing power Π, in the diagonal, isoelectronic cations from Group 1A to 3A, shown in Table (7.4).
From these calculated values, the purely electronic parameters, η and χ, do not seem to follow a consistent Diagonal Rule in any Sequence. On the other hand, the spatio-electronic parameters, α and Π, do display approximately constant values, especially in the Sequences of the heavier Elements. From these correlations , it appears that the strength and the type of bonds formed by these cations relies on their polarizing power Π, as electron acceptors and on their polarizability α, as electron donors. In theoretical terms, this need for spatio-electronic information means that the model must describe the bonding energies available at the actual radii of the bonding ions.
Heavy Group 3A Element Chemistry
When this spatio-electronic bonding model is used to describe the chemical behaviour of the heavier Elements of Group 3A, it cab be used to understand and predict both their electronic structures and thier patterns of behaviour.
Table.4 Diagonal Sequences for Maximum Oxidation Number Cations
| Sequence of Isoelectronic Maximum SON Cations | Electronic
Property |
Strength of Property (MKS units) | ||
| Group 1A | Group 2A | Group 3A | ||
| H +I ⇒ Be +II ⇒ Al +III | η | 675 | 6500 | 4400 |
| α | 3 x 10-10 | 0.0026 | 0.1 | |
| χ | ≈8000 | 8250 | 7100 | |
| Π | ≈200 | 33 | 13.5 | |
| Li +I ⇒ Mg +II ⇒ Ga +III | η | 3375 | 3150 | 1600 |
| α | 0.2 | 0.21 | 0.45 | |
| χ | 3850 | 4600 | 4500 | |
| Π | 6.4 | 5.25 | 7.3 | |
| Na +I ⇒ Ca +II ⇒ In +III | η | 2025 | 1850 | 1250 |
| α | 1.5 | 1.8 | 1.23 | |
| χ | 2525 | 3000 | 4000 | |
| Π | 2.5 | 1.85 | 1.6 | |
| K +I ⇒ Sr +II ⇒ Tl +III | η | 1300 | 1550 | 500 |
| α | 6.35 | 3 | 4.1 | |
| χ | 1725 | 2650 | 2500 | |
| Π | 1.25 | 1.35 | 0.6 | |
| Rb +I ⇒ Ba +II | η | 1125 | 1450 | - |
| α | 9 | 6 | - | |
| χ | 1525 | 2400 | - | |
| Π | 1 | 1.08 | - | |
The Group3A Halides
The neutral molecule AlCl3 would be formally electron deficient because, as in the BF3 molecule, the three bonding pairs donated by the chloride ions shared pairs could not fill the stable octet of the n = 3 valence shell, as shown in Figure 6
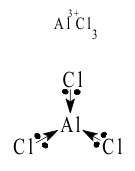
Thus, in Lewis acid-base terms, AlCl3 is an acid, seeking the donation of the necessary additional electron pair from a Lewis base to complete its valence shell “octet” of electrons. However, the polarizing power of Al +III is much lower than that of B +III and cannot stabilize the same strong Lewis acid-base complexes with Hard or non-polarizable bases. From the Diagonal Rule, Al +III does resemble Be +II and forms complexes with Softer, more polarizable bases. This includes the chloride ion and allows the equilibrated “dimerization” of two units ;
2 AlCl3 Û Al2Cl6 (10)
by the Lewis base donation of non-bonding chloride ion pairs to form bridge bonds, shown in Figure 7
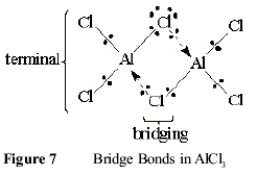
The dimer obeys the Octet Rule at each Al +III ion because their valence shells mimic the rare gas structure of [Ar]. These shells are filled by two pairs of electrons from bonds with “terminal” chloride ions and with two pairs shared in bridge bonds
The Group 3A Oxides
Using the Diagonal Rule, the water chemistry of the Group 3A Elements can be predicted by comparing it to that of the earlier members of each Sequence. Thus, from Table (7.4), the polarizing power of Al +III suggests that it could hydrolyse neutral water like Be +II and that Ga +III would be able to hydrolyse it like Mg +II. The lower polarizing power of the heavier Element cations In +III and Tl +III indicates that they could not hydrolyse neutral water but instead would form simple aquated cation complexes like Ca +II and Sr +II ions.
These predictions are supported by the empirical Activities of the ions in this Group. In strong acid, all of these ions form the “hexaquo” complex, such as the [Al (H2O )63+] complex ion
However, as the pH is raised, the ions respond as Bronstead acids and progressively lose Hydrogen ions because the negative charge on the bound oxygen is polarized away from the (O-H) bond, while the external H2O molecules or (OH-) ions provide alternative bonding sites, as shown in Figure 8.
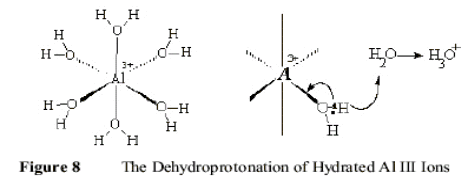
This process continues as the pH is raised and further Hydrogen ions are lost until the neutral, hydrated oxide [Al(OH)3(H2O)3]0 is formed as an gelatinous, insoluble polymeric solid. The pH at which this happens depends on the polarizing power of the Group III ion, from pH 6 with Al +III to pH 11 for Tl +III. At higher pH levels, these neutral hydrous oxides begin to respond as sources of Hydronium ions, [H3O+] so that at high pH, Al-O bonds are being broken as these aquated protons are released. This process of dehydration eventually results in formation of the dry oxide, Al2O3