
Group 2A; The Hydrolysing Cations
Overall Relationships of Structures to Activities
The Elements of Group 2A begin in the second row of the Periodic Table because the filling of the (1s)2 orbital of Helium completes the first row of the Table, as shown in Figure 1. All Group 2A Elements have electronic configurations [Rare Gas ](ns)2 with n from 2 to 7.
Spatial and Electronic Structures of the Elements
The neutral atom configuration of these Elements is the filled (ns)2 electronic Structure in which these two electrons occupy an s orbital outside the core of the filled p orbital. As in Group 1A, these (ns)2 electrons can penetrate this core from all directions but again, the core of filled p orbitals provides a shield with no defects which makes this valence pair of electrons very unstable. This shielding is least effective for Beryllium but becomes more dense and difficult to penetrate down the Group.
The resulting decrease in Effective Nuclear Charge, ZEff is shown clearly by the progressive increase in the atomic radius, representing the spatial aspect of atomic Structure of and the decrease in Hardness and Electronegativity, representing the electronic aspects of atomic Structure, from Beryllium down to Radium The full correlation of these electronic configurations to the corresponding spatial and electronic Structures is shown in Table 1
Table 1 Group 2A Correlation of Electron Configuration to Electronic Structure
|
Element Name |
Structural Configuration [core electrons] (valence electrons) |
r nm |
η kJ/M |
χ kJ/M |
|
Beryllium |
[(1s)2] (2s)2 |
0.112 |
460 |
440 |
|
Magnesium |
[(1s)2(2s)2(2p)6] (3s)2 |
0.16 |
395 |
340 |
|
Calcium |
[(1s)2(2s)2(2p)6(3s)2(3p)6] (4s)2 |
0.197 |
295 |
295 |
|
Strontium |
[(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)10(4p)6] (5s)2 |
0.215 |
275 |
275 |
|
Barium |
[(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)10(4p)6(5s)2 (4d)10(5p)6] (6s)2 |
0.224 |
250 |
250 |
|
Radium |
[(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)10(4p)6(5s)2 (4d)10(5p)6(6s)2(4f)14(5d)10(6p)6] (7s)2 |
? |
255 |
255 |
Oxidation of the Elements
As in Group 1A, the Hardness and Electronegativity of Group 2A Elements begin at moderate values in Beryllium and decrease down the Group to Radium. These Periodic trends show that the Electronegativity of most Group 2A Elements, is less than that of most nonmetallic Elements. In a reaction between these two types of Element, Pauling’s Electroneutrality Rule predicts that to minimize the energy of the whole system, the Group 2A atoms lose their (ns)2 electrons to the non-metallic atom. This “oxidizes” the Group 2A Element to a stable cation with Oxidation Number +II.
At the same time, by acquiring the valence electrons of the Group 2A atom, the nonmetallic atom is “reduced” to a negative anion. For example, in reaction with molecular
Halogens, X2 ;
M + X2 ⇒ MX2 (1)
The resulting anion can then form any type of chemical bond to the Group 2A cation.
Acid-Base Chemistry of the Cations, M +II
The type of [ M-X ] bond formed in each case, covalent, donor covalent or ionic, depends on the relative polarizing powers Π and polarizabilities α of the two Elements forming the cation-anion pair. Using the definitions in equations (R2.1) and (R2.2), the values of all these Structural properties for the stable Oxidation Number +II of this Group are given in Table (2).
Table 2 Electronic Structures of Stable Oxidation Numbers of Group 2A Cations
|
Stable ON |
Configuration [Core](valence) |
r nm |
η kJ/M |
(106)α (nm3)M/kJ |
χ kJ/M |
Π (10-4) kJ/Mnm |
|
Be +II |
[ He ](2s)0 |
0.025 |
6500 |
0.0026 |
8250 |
33 |
|
Mg +II |
[ Ne ](3s)0 |
0.06 |
3150 |
0.21 |
4600 |
5.25 |
|
Ca +II |
[ Ar ](4s)0 |
0.101 |
1850 |
1.8 |
3000 |
1.85 |
|
Sr +II |
[ Kr ](5s)0 |
0.115 |
1550 |
3 |
2650 |
1.35 |
|
Ba +II |
[ Xe ](6s)0 |
0.135 |
1450 |
6 |
2400 |
1.08 |
|
Ra +II |
[ Rn }(7s)0 |
? |
≈1400 |
>9.0 |
≈2200 |
<1.00 |
Covalent Bonding
From Table (4.2), the first two Elements of this Group apparently have high enough values of Hardness and Electronegativity to permit them to share rather than donate their valence electrons. By the Diagonal Rule, these Elements respectively resemble Hydrogen and Lithium and likewise form covalent bonds to Hard Elements such as Carbon. This is shown by formation of the linear (straight) molecule “dimethylberyllium”, in which the high polarizing power of Be +II forces the carbon to share its valence electrons in a covalent bond ;
H3C-Be-CH3 .
Similar bonding conditions exist in the organomagnesium “Grignard Reagents”, including
methylmagnesium iodide;
H3C-Mg-I ,
and in the “Sandwich” compound, dicyclopentadienylmagnesium ;
H5C5-Mg-C5H5
Donor Covalent Bonding to Group 2A Cations
In comparison with the Group 1A elements, the higher Hardness and Electronegativity of Group 2A Elements promotes the formation of Donor covalent bonds instead of simple ionic bonds with most anions. The cations act as Lewis acids, accepting charge from Lewis bases such as water. The strength of the donor covalent bond depends on both the Polarizing Power of the Lewis acid, in this case the cation and the Polarizability of the Lewis base, in this case the water molecule. The more charge is donated, the stronger is the resulting donor covalent bond shown in Figure 2.
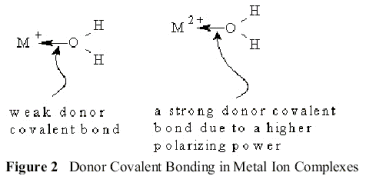
In Be2+O2-, the polarizing power of Be2+ is high enough to polarize the electron pairs of the oxide ion into a donor covalent bond. As shown in Figure (6.3), the Lewis acid base complex is ;
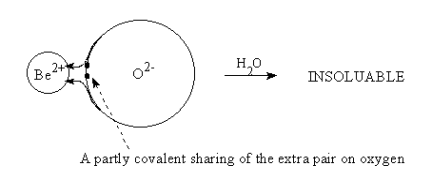 Figure 3 Donation of Electron Density in a
Lewis Acid Base Complex
Figure 3 Donation of Electron Density in a
Lewis Acid Base ComplexThis type of bonding can be described as a distortion of the HOMO orbitals of the basic oxide ion, in which one of the electron pairs in the filled (2p)6 orbital is distorted by the Be2+ ion into a donor pair, as shown in Figure 4.
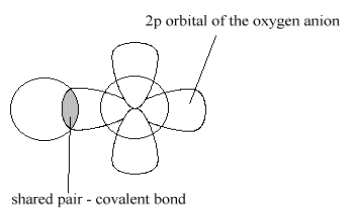 Figure 4 Donor Covalent Bonding due to high
polarizing power of Be2+
cation
Figure 4 Donor Covalent Bonding due to high
polarizing power of Be2+
cationThis is shared into the empty Lewis acid (2s) orbital but is not permanently given back to form a neutral Be atom. This reversibility distinguishes it from the irreversible covalent bond, in which the pair is permanently shared between bond partners
Hydrolysis of Group 2A Cations
However, in compounds of cations with very high polarizing power, the donor covalent bonds show many of the same properties seen with true covalent bonds. The high polarizing power pulls so much of the anionic charge into the localized bond that the outside of the Lewis complex appears to be neutral in charge. This means that the physical properties of the compound resemble those of truly covalent neutral compounds with low melting points and low solubilities in polar solvents. For example, in solid phase BeO, the charge requirement of the Be +II ion is mostly satisfied internally by polarization of the negative oxide charge. This leaves only a small excess positive charge on the Be atom and an equally small negative charge on the O atom, as shown in Figure 5
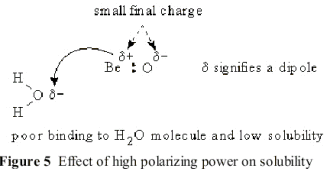
However, this small remaining dipole allows an attack by the negative end of the water dipole on the Be +II cation. In this Transition State, this interaction splits the water molecule into a Hydrogen ion and a hydroxide ion, as shown in Figure .6.
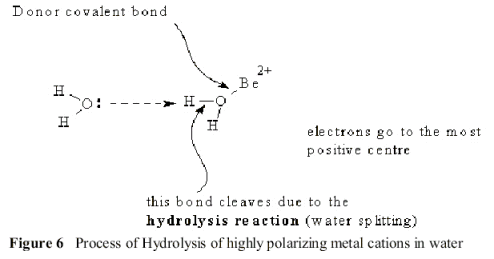
The Hydroxide binds to Be +II cation and Hydrogen ion is transferred to the oxide ion, forming a gelatinous, insoluble hydroxide at equilibrium:
BeO + H2O Û [Be2+(OH-)2 ] (2)
Acid Base Complex
Gelatinous Hydroxides of Group 2A Cations
The high polarizing power of these Group 2A ions makes the hydrated oxide, [Be(OH)2] neutral in apparent charge and consequently insoluble in polar solvents, especially water. However, the exact degree of hydration is uncertain and the resulting insoluble material forms as a gelatinous deposit of uncertain composition with no crystalline Structure. This means that in any added Bronstead acid or base initially comes in contact with the surface of the gelatinous, hydrated oxide and must penetrate through this medium in order to react with the Be-O bonds. This penetration process often requires a significant length of time, so while it is occurring, the yield of the reaction is dominated by the rate at which the acid or base penetrates, rather than by the position of the final equilibrium on the Reaction Quotient.
Amphoteric Soluble Complexes of Group 2A Cations
The full range of Group 2A acid base chemistry is illustrated best by the Activities of these Beryllium compounds in water. The shared charge in donor covalent bonds from oxygen, as oxide in BeO or hydroxide in Be(OH)2 , can be separated in two opposing ways. One is by adding a Bronstead acid to the liquid phase to force the neutral hydrated metal oxide to act as a base ;
H3O+ + [Be(OH) 2] Û Be(OH)(OH2)+ + H2O (3a)
Acid Base Conjugate Acid Conjugate Base
This can be quantitated with its acid hydrolysis equilibrium constant ;
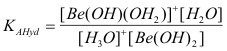
(3b)
The second is to add a Bronstead base and force the hydrated oxide to act as an acid ;
[Be(OH)2] + OH- Û Be(OH)(O)- + H2O (4a)
Acid Base Conjugate Base Conjugate Acid
which can be quantitated with the corresponding base hydrolysis constant ;
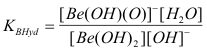
(4b)
The combination of these two reactions shows that hydrated BeO can dissolve in water as a cationic species at low pH and equally well, as an anionic species at high pH. This dual solubility occurs with most Group II cations in water and is called “amphoterism” or amphoteric behaviour. The hydrolysis constants increase down the Group, with decreasing Hardness, until at Radium, the cation is soluble, even in neutral water, resembling its Diagonally related Group 1A cation, Cesium.
Solubility of Group 2A Cations From Salts
Since the hydroxide of Group 2A cations are insoluble in neutral water, it may be expected that their salts with other, equally Hard anions may be equally insoluble. To assess this, the stability of solvated Group 2A cations must be compared to the stability available in the solid phase salts of these anions. In contact with water, these solids can react to produce dissolved, “aquated” species;
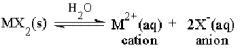
(5a)
in which the forward reaction represents dissolution and the back reaction represents precipitation. In solution the cations form solvated, in this case, hydrated complexes shown in Figure 7;
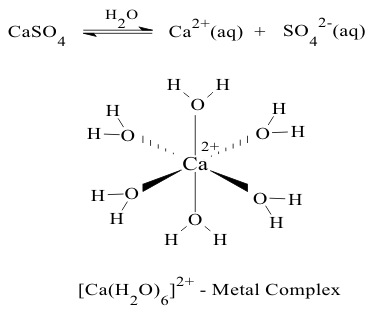 Figure 7 The Hydrated Calcium 2A
Cation
Figure 7 The Hydrated Calcium 2A
CationThis equilibrium can again be quantitated by calculating the appropriate equilibrium constant, which, in this case is called the “solubility product” of the salt. In this type of equilibrium between solid and liquid forms of the reagent, the concentration of the solid [MX2]s in itself is arbitrarily set to 1.0M in the pure solid ;
(5b)
so that the constant simplifies to;
KSP = [M(aq)2+][X(aq)-]2 (5c)
At first, the exergonic forward reaction dominates but as the water penetrates the solid, the forward and back reaction rates become equal and the solution becomes saturated. At this point on the Reaction Quotient, Ksp can be used to determine the concentration of CaSO4 in the saturated solution. If this analysis is applied to the solubility of CaSO4, the balanced chemical reaction is ;
CaSO4 Û Ca2+ + SO42- (6a)
so that in solution ; [Ca2+] = [SO42-] (6b)
In these conditions, the solubility of the salt is defined by the value of either [Ca2+] or [SO42-]and can be calculated, as with any equilibrated reaction by the two step approximation :
[CaSO4] Û [Ca2+ ] + [ SO42-] (6c)
Before equilibrium 1.0 0 0
After equilibrium 1.0 x x
and then substituting these values into the equilibrium to produce ;
Ksp = x2 = 1.3x10-8
=
![]() » 1.5x10-21
mol/L (6d)
» 1.5x10-21
mol/L (6d)
Similarly for the salt CaF2, the balanced chemical equation is ;
CaF2 Û Ca2+ + 2F - (7a)
so that in solution [Ca2+] = 2 [F - ] (7b)
Under these conditions, the solubility of the salt is defined by the value of either [Ca2+] or [SO42-]and can be calculated, as with any equilibrated reaction by the two step approximation :
[CaSO4] Û [Ca2+ ] + [F - ] (7c)
Before equilibrium 1.0 0 0
After equilibrium 1.0 x 2x
and then substituting these values into the equilibrium to produce ;
KSP = (x)(2x)2 = 4x3 = 4.0 x 10 -11(7d)
x = ( 10 x 10 -12 ) 1/3 ≈ 2.2x10-4
If the equilibrium is approached from the back reaction, a ‘supersaturated solution must be stimulated to precipitate the crystallised solid to form the equilibrated saturated solution. This is done by catalysing precipitation with “nucleating agents” which “seed” precipitation by mimicking the crystals of the desired solid. The process is “propagated” on the surface of the seed crystal.
Ionic Bonding
From this description of bonding by Group 2A cations, it is clear that the purely ionic bonding exhibited by Group 2A cations is rare. The Diagonal Rule correlates the Structures formed by Beryllium back to those of Hydrogen and by Magnesium back to those of Lithium. In this way, this Rule predicts that only the cations of the heavier Elements, from Calcium down the Group to Radium form ionic solid-state bonds with “lattice” Structures in which the positive charge on the cation exerts a purely electrostatic, Coulombic attraction on anions.